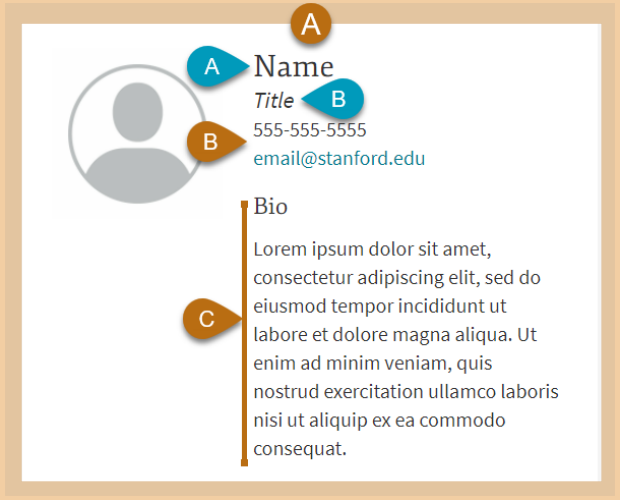
Profile (CAP)
Import Stanford Profiles (CAP) with photos, bios contact info. and more
Profile components import and synchronize CAP user data to create individual or lists of staff entries. Keeping all data in one place eases information management and ensures data accuracy.
Features
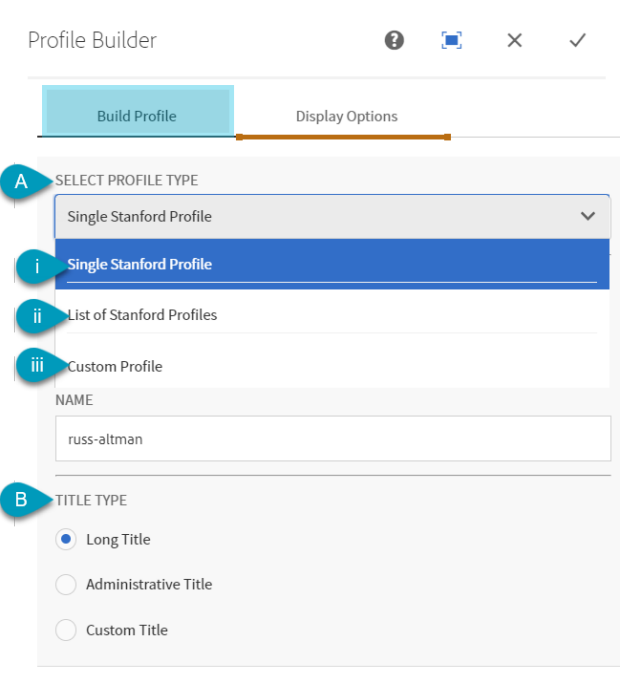
- Display a single or list of profiles
- Custom (manual) option for those without a profile
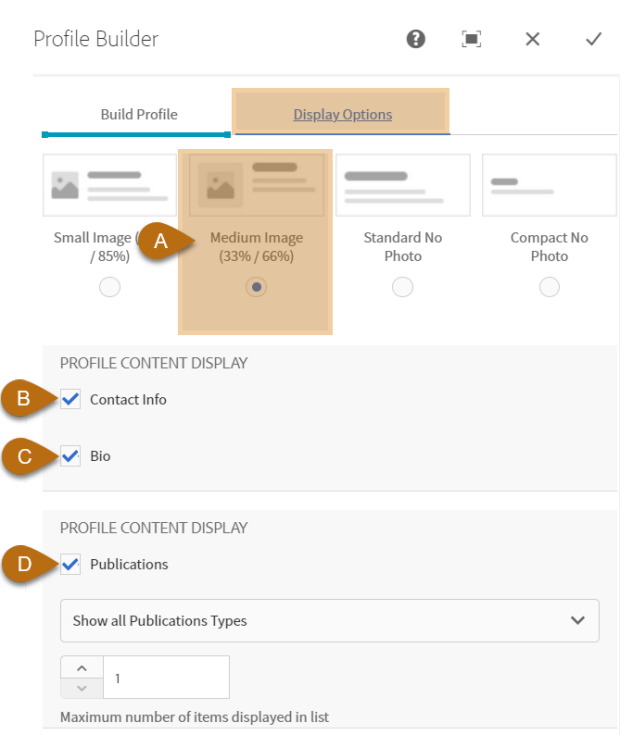
- Display toggles for bios, contact info and titles
- Publication option for single profiles
- Multiple display options to fill different spaces
Pro Tips
- CAP profiles must be set to public to import
- Custom & Single Profile options are one entry per component
- Use Single Profile for the publications feature
Visual Manual
Align left-column colors with right-column content
Output Key
Displays the input fields