Neurodegeneration Research in the Südhof Lab: A Focus on Synapses
The last decade has arguably been the most insightful for neurodegeneration research since the vibrant period of gene discovery in the 1990’ies. To mention just a few major advances, large-scale GWAS studies identified scores of genetic variants associated with neurodegenerative diseases, cryo-EM experiments showed that protein aggregates found in neurodegenerative disorders universally exhibit cross-beta structures, and transcriptomics combined with human genetics uncovered a major role for microglia in neurodegenerative disorders. Moreover, the first disease-modifying therapy was achieved in Alzheimer’s disease (AD) with Ab-antibodies, although patient benefits were modest.
Despite these great advances, however, we still have only a rudimentary understanding of why synapses are lost and why neurons die in neurodegenerative diseases in general and in AD in particular. Solving the pathogenesis of AD is a generational challenge. The brains of many, maybe all, AD patients not only feature Ab-containing plaques and tau-containing fibrillary tangles, but also often contain not only aggregates composed of Ab peptides (plaques) and tau (tangles), but also additional aggregates formed by synuclein (Lewy bodies), TDP43, and TMEM106b. Moreover, a robust inflammatory response is invariably observed that may reflect a defense mechanism.
Our lab’s work on neurodegeneration is focused on the role of synapses in AD. This focus is motivated by the robust evidence implicating synapses in neurodegenerative disorders, leading us to attempt to leverage our expertise in analyzing synapses for insight into AD pathogenesis. This focus is not meant to imply that other components of AD pathogenesis are unimportant. In contrary, neuroinflammation is crucial and other processes associated with protein aggregates also likely contribute.
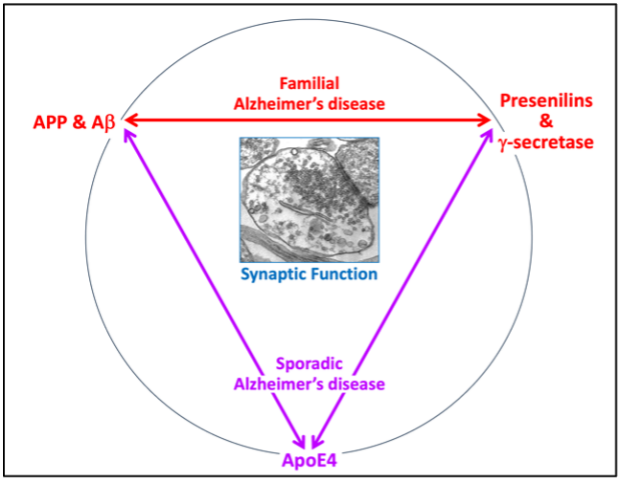
Our work on synaptic dysfunction in neurodegeneration has concentrated on the specific role in synapses of AD genes that exhibit the biggest effect sizes, namely on the familial AD genes encoding APP and presenilins and the sporadic AD gene ApoE4. Our overall hypothesis is that these three genes perform, among others, a role in maintaining synaptic integrity, such that their dysfunction puts the maintenance of synapses at risk, as schematically illustrated on the right. The role of these genes does not necessarily operate in the same pathways although some overlap seems likely.
APPSwedish Familial Alzheimer’s disease mutation enhances synaptic connectivity
APP point mutations as well as APP gene triplications increase Ab production and/or aggregation and cause AD, suggesting that Ab is an initiator of AD pathogenesis1. In thousands of papers, Ab has been shown to be toxic for neurons. More than a dozen of Ab-receptors were published to explain Ab toxicity. Collectively these studies led to the notion that AD is caused by toxic Ab damaging synapses and neurons2. However, most of these studies were carried out in rodents and involved overexpression of mutant APP, addition of exogenous Ab, or cell lines with distinct genetic backgrounds. Given that, in familial AD symptoms only develop after decades of overproduction of Ab, it seems likely that Ab induces AD by more complex non-linear pathways than direct toxicity.
To revisit the question of Ab toxicity, we recently engineered a conditional heterozygous APP point mutation that replicates a familial AD mutation (the so-called ‘Swedish’ mutation) into human neurons from ES and iPS cells3. In human neurons derived from the engineered ES/iPS cells, Cre-recombinase induces the APPSwedish mutation whereas Flp-recombinase induces the APPWT allele. In parallel, we created human neurons carrying conditional APP deletions. These engineered neurons allowed us to analyze the effect of the Swedish mutation and APP deletion on human neurons under precisely controlled, physiological conditions with identical genetic backgrounds in control and mutant neurons.
Strikingly, we found that the APPSwedish mutation induced an increase in synaptic connectivity instead of an impairment, even though Ab production was increased as expected. In contrast, the APP deletion caused a loss of synapses. The effect of the APPSwedish mutation was blocked by a BACE1 inhibitor, indicating that the APPSwedish mutation enhances synapse formation by elevating Ab production. These results suggest that, instead of being deleterious, the increased production of Ab enhances synapse formation. Thus, our results indicate that AD does not simply develop as a result of Ab toxicity but is caused by an indirect effect of Ab.
Knowledge gaps. We do not know how Ab functions in synapse formation and why the increased Ab secretion in familial AD leads to neurodegeneration. One hypothesis is that chronic overexcitement of neurons by Ab might be deleterious in the long run. An alternative hypothesis is that increased production of Ab causes increased aggregation, which in turn sucks up all the free Ab, thereby rendering AD an Ab loss-of-function disease. The latter hypothesis is consistent with the finding that a decrease in free Ab in the CSF is uniformly observed in both familial and sporadic AD4.
References
1. Tcw, J., Goate A.M. (2017) Genetics of β-Amyloid Precursor Protein in Alzheimer's Disease. Cold Spring Harb. Perspect. Med. 7, a024539.
2. Mucke, L., Selkoe, D.J. (2012) Neurotoxicity of amyloid β-protein: synaptic and network dysfunction. Cold Spring Harb. Perspect. Med. 2, a006338.
3. Zhou, B., Lu, J.G., Siddu, A., Wernig, M., Südhof, T.C. (2022) Synaptogenic Effect of APP-Swedish Mutation in Familial Alzheimer’s Disease. Science Transl. Medicine 14, eabn9380.
4. Rosén, C., Hansson, O., Blennow, K., Zetterberg, H. (2013) Fluid biomarkers in Alzheimer's disease - current concepts. Mol. Neurodegener. 8, 20.
Suppression of g-secretase activity lowers cholesterol levels in neurons but not glia
Presenilin-1 and -2 mutations cause familial AD similar to APP mutations1. Presenilins are g-secretase subunits; their mutations induce a decrease in g-secretase activity and alter Ab production1-3. However, mutations in other g-secretase subunits that also reduce g-secretase activity do not cause familial AD but hidradenitis suppurativa, a skin disease4, suggesting that a decrease in g-secretase activity alone does not cause AD.
We examined the function of g-secretase activity in human neurons by testing the effect of chronic pharmacologic g-secretase inhibition or of a deletion of presenilin-1. Both manipulations caused a large decrease in the presynaptic neurotransmitter release probability5 consistent with our earlier mouse data6. RNAseq analyses revealed that chronic g-secretase inhibition caused a massive upregulation in human neurons but not in co-cultured mouse glia of genes mediated cholesterol synthesis and cholesterol transport5. Direct cholesterol measurements showed that neuronal but not glial cholesterol levels were decreased, suggesting that the increase in cholesterol synthesis and transport gene expression was an indirect reaction to the decrease in cholesterol levels5. Thus, in neurons but not in glia, g-secretase is essential for maintaining normal cholesterol levels. Moreover, statins that also lower neuronal cholesterol levels produced the same decrease in presynaptic release probability as the chronic suppression of g-secretase activity, suggesting that the decrease in release probability produced by chronic g-secretase suppression was a direct consequence of the decrease in cholesterol levels5.
Several AD-associated genes, most prominently ApoE4, are thought to function in cholesterol metabolism7, suggesting that our findings could potentially be relevant for AD pathogenesis. However, the major value of our findings is in identifying a novel function for g-secretase in neuronal cholesterol metabolism. Please note that neurons are unusual in terms of cholesterol metabolism since neurons are the only cells in the body that metabolize cholesterol to 24-hydroxy-cholesterol for secretion, suggesting there may be a link8.
Knowledge gaps. It is unknown how g-secretase regulates cholesterol levels and whether this regulation is related to APP, Ab, and AD pathogenesis. Moreover, it is unclear why the role of g-secretase in cholesterol metabolism is neuron-specific and if this role is related to the neuron-specific degradation of cholesterol by 24-hydroxylation.
References
1. Hur, J.Y. (2022) γ-Secretase in Alzheimer's disease. Exp. Mol. Med. 54, 433-446.
2. Sun, L., Zhou, R., Yang, G., Shi, Y. (2017) Analysis of 138 pathogenic mutations in presenilin-1 on the in vitro production of Aβ42 and Aβ40 peptides by γ-secretase. Proc. Natl. Acad. Sci. USA. 114, E476-E485.
3. Kelleher, R.J. 3rd, Shen, J. (2017) Presenilin-1 mutations and Alzheimer's disease. Proc. Natl. Acad. Sci. USA. 114, 629-631.
4. Mintoff, D., Pace, N.P., Borg, I. (2022) Interpreting the spectrum of gamma-secretase complex missense variation in the context of hidradenitis suppurativa – An in-silico study. Front. Genet. 13, 962449.
5. Essayan-Perez, S., Südhof, T.C. (2023) Neuronal gamma-secretase regulates lipid metabolism, linking cholesterol to synaptic dysfunction in Alzheimer’s disease. Neuron 111, 3176-3194.
6. Zhang, C., Wu, B., Beglopoulos, V., Wines-Samuelson, M., Zhang, D., Dragatsis, I., Südhof, T.C., and Shen, J. (2009) Presenilins are essential for regulating neurotransmitter release. Nature 460, 632-636.
7. Dai, L., Zou, L., Meng, L., Qiang, G., Yan, M., Zhang, Z. (2021) Cholesterol Metabolism in Neurodegenerative Diseases: Molecular Mechanisms and Therapeutic Targets. Mol. Neurobiol. 58, 2183-2201.
8. Moutinho, M., Nunes, M.J., Rodrigues, E. (2016) Cholesterol 24-hydroxylase: Brain cholesterol metabolism and beyond. Biochim. Biophys. Acta 1861(12 Pt A), 1911-1920.
ApoE boosts synapse formation in an ApoE4>ApoE3>ApoE2 rank potency order
In early studies, we showed in mice that ApoE enhances neurodegeneration induced by a-synuclein overexpression1, a finding that has been reproduced by others and expanded to the demonstration that ApoE4 is more potent than ApoE2 and ApoE3 in promoting neurodegeneration induced by a-synuclein or mutant tau2,3. Thus, ApoE promotes neurodegeneration such that ApoE4 is more potent than ApoE2 or ApoE3, consistent with their relative association with AD4.
We were therefore surprised to find that exogenous ApoE increases synapse numbers and synaptic connectivity in cultured human neurons and that ApoE4 was more potent in this effect than ApoE3, which in turn was more potent than ApoE25,6. The ApoE-dependent enhancement of synapse formation was mediated by a classical ApoE receptor that could be blocked with RAP, a protein-inhibitor binding to such receptors. The increase in synapse numbers involved activation of a general MAP-kinase signaling pathway that, among others, increased APP transcription5,6. Other laboratories questioned these results, suggesting that in our original experiments we may have used non-lipidated ApoE that could have produced artifacts7. We therefore performed independent validation experiments using astrocyte-produced ApoE and native human LDL particles containing ApoE, which confirmed the original results and revealed a more general signaling activation than we had originally reported6.
Knowledge gaps. No currently available data provide conclusive evidence for a particular function of ApoE in brain, different from its assured role in the peripheral circulation where ApoE acts as a lipid transport apo-protein8. The fact that ApoE in combination with TREM2 is linked to accumulation of lipid droplets in microglia9,10 suggests that ApoE also acts in cholesterol transport in brain, but no actual lipid transport was demonstrated and the effects could be indirect. Our data suggest a signaling role but again the phosphorylation events we observed could represent an activated metabolic state instead of a direct signal. A third possibility is that ApoE regulates reelin signaling, which is raised by the finding that a reelin point mutation appears to protect against AD caused by a presenilin mutation11 and that reelin receptors are also ApoE receptors in which ApoE competes with reelin for binding12. Thus, no definitive conclusions about ApoE function in brain are currently possible and establishing the basic function of ApoE in brain is probably the most pressing current challenge.
It is noteworthy that in brain, ApoE is a major injury-response gene that is activated in astrocytes and microglia in response to pathology. ApoE deletions in mice appear to induce no major brain phenotype, suggesting that the basic function of ApoE in brain is non-essential under non-pathological conditions. Addressing the knowledge gap of the basic ApoE function in brain will be required for development of anti-ApoE4 therapies, which may be a promising avenue to AD drugs but are currently rendered difficult by the essential role of ApoE in blood lipid transport.
References
1. Gallardo, G., Schlüter, O.M., Südhof, T.C. (2008) A molecular pathway of neurodegeneration linking a-synuclein to ApoE and Ab-peptides. Nature Neurosci. 11, 301-308.
2. Davis AA, et al. (2020) APOE genotype regulates pathology and disease progression in synucleinopathy. Sci. Transl. Med. 12, eaay3069.
3. Shi, Y., et al. (2017) ApoE4 markedly exacerbates tau-mediated neurodegeneration in a mouse model of tauopathy. Nature 549, 523-527.
4. Lou T, Tao B, Chen M. (2023) Relationship of Apolipoprotein E with Alzheimer's Disease and Other Neurological Disorders: An Updated Review. Neuroscience 514, 123-140.
5. Huang, Y.A., Zhou, B., Wernig, M., Südhof, T.C. (2017) ApoE2, ApoE3, and ApoE4 Differentially Stimulate APP Transcription and Ab Secretion. Cell 168, 427-441.
6. Huang, Y.A., Zhou, B., Nabet, A.M., Wernig, M., Südhof, T.C. (2019) Differential Signaling Mediated by ApoE2, ApoE3, and ApoE4 in Human Neurons Parallels Alzheimer's Disease Risk. J. Neurosci. 39, 7408-7427.
7. Zhao, J., et al. (2020) APOE4 exacerbates synapse loss and neurodegeneration in Alzheimer's disease patient iPSC-derived cerebral organoids. Nat. Commun. 11, 5540.
8. Goldstein, J.L., Brown, M.S. (2015) A century of cholesterol and coronaries: from plaques to genes to statins. Cell 161, 161-172.
9. Tcw J, et al. (2022) Cholesterol and matrisome pathways dysregulated in astrocytes and microglia. Cell 185, 2213-2233
10. Nugent AA, et al. (2020) TREM2 Regulates Microglial Cholesterol Metabolism upon Chronic Phagocytic Challenge. Neuron 105, 837-854
11. Lopera F, et al. (2023) Resilience to autosomal dominant Alzheimer's disease in a Reelin-COLBOS heterozygous man. Nature Med. 29, 1243-1252.
12. Alexander A, Herz J, Calvier L. (2023) Reelin through the years: From brain development to inflammation. Cell Rep. 42, 112669.
Continuing puzzles: Where do we go from here?
Our results on the APPSwedish mutation and on ApoE4 appear to be counterintuitive and are mechanistically unexplained. Thus, the most immediate need is to validate these results and to identify their molecular basis. In addition, our results on g-secretase suggesting a neuron-specific role in regulating cholesterol levels is also mechanistically enigmatic, albeint intriguing, given that neurons are known to exhibit an unusual cholesterol metabolism. Here, the need seems pressing to determine whether the neuron-specific role of g-secretase in maintaining cholesterol levels is related to the neuron-specific 24-hydroxylation of cholesterol.
With such mechanistic studies addressing specific questions, we hope to also clarify a more general important question: How to synthesize these results into a coherent, mechanistically plausible conceptual framework that pulls them together and relates them to AD pathogenesis. Key to such a conceptual framework is the time course of AD pathogenesis. Even familial mutations in APP and presenilin genes that invariably cause AD require decades to produce the disease. Thus, these mutations must have a cumulative mechanism of action. The notion that the APP and presenilin mutations, and ApoE4 as the most important genetic risk factor for sporadic AD, act by a common pathway is strongly supported by the finding that a point mutation in ApoE (the so-called ‘Christchurch’ mutation) delays the onset of familial AD caused by a presenilin mutation by decades1 and that ApoE is a cholesterol-transport protein that may also act as such in brain. What precise mechanisms connect these ‘dots’, however, remains to be established.
References
1. Arboleda-Velasquez, J.F., et al. (2019) Resistance to autosomal dominant Alzheimer's disease in an APOE3 Christchurch homozygote: a case report. Nature Med. 25, 1680-1683.