The Basics of CF
What Is CF?
Cystic Fibrosis (CF) is one of the most common genetic (inherited) diseases in America. It is also one of the most serious. It mainly affects the lungs and the digestive systems in the body, causing breathing problems and problems digesting foods. It is a chronic disease that currently has no cure.
What Happens?
Glands in the body that usually produce thin, slippery secretions (like sweat, mucous, tears, saliva, or digestive juices) produce thick, sticky secretions. These thick, sticky secretions plug up the ducts (small tubes) that should carry the secretions either outside of the body or into a hollow organ such as the lungs or the intestines. This can affect vital body functions such as breathing or digestion.
Why?
CF is present at birth because both parents carried a CF gene, and their infant inherited a CF gene from each parent. Not every child from this family will necessarily have CF. Other children could inherit a single CF gene from just one parent, and thus become a carrier for CF, or they could inherit no CF gene and be completely free from CF. Since 1989, when the CF gene was first discovered, research has made great progress in understanding CF.
How is CF diagnosed?
A suspicion of CF occurs when some of these symptoms are present:
- Persistent cough, wheezing, or recurrent pneumonia
- Good appetite, but poor weight gain
- Loose, bad-smelling bowel movements
- A salty taste to the skin
- Clubbing (enlarging) of the fingertips
A simple, painless test called a sweat chloride test can then be done. CF causes a large amount of salt to be lost in the sweat. Measuring the amount of salt in the sweat can determine whether or not a person has CF.
Genetics and CF
What is a gene?
A gene is the basic unit of heredity. Genes are responsible for the physical characteristics that each person has (like eye color, facial features, and many health conditions). Each gene occupies a certain location on a chromosome (a thread-like material that is located in the nucleus of every single cell in the body). Chromosomes come in 23 pairs, and each chromosome carries thousands of genes.
What happens?
Each gene has a specific role in determining how a person's body is put together and how it functions. The role of a gene is determined by its individual DNA code (deoxyribonucleic acid, the chemical coding for a gene). DNA is made up of four building blocks called bases. These bases are joined in a specific order for each gene. When a change occurs in the arrangement of the bases, it can cause the gene not to work properly.
What are genetic disorders?
A structural gene change which can cause a disease or a birth defect is called a mutation. Genes are inherited in pairs, with one gene inherited from each parent to make the pair. Cystic fibrosis occurs when both genes in the pair have a mutation. A person with cystic fibrosis inherits one CF gene from each parent. Cystic fibrosis is a genetic disorder caused by inheriting a pair of genes that are mutated or not working properly.
The Cystic Fibrosis Gene
Everyone inherits two copies of the CFTR (cystic fibrosis transmembrane conductance regulator) gene. However, some of the inherited copies are mutations. To date, over 700 mutations of the CFTR gene have been identified. A person with CF inherits two mutated copies of the CFTR gene. These mutations can either be homozygous, the same, or heterozygous, different mutations. The most common mutation is delta F508, accounting for approximately 70% of all mutations. Those homozygous for this mutation tend to be pancreatic insufficient.
What Does the Mutation Do?
The CFTR gene is a protein that functions as a chloride channel. A chloride channel helps maintain the proper balance of salt and water within a cell. A mutation in CFTR causes a dysfunction of the salt and water balance. This causes dehydration of the secretions (thick mucous) and excessive loss of salt in sweat.
What is a carrier?
A carrier is a person who only has one copy of the mutated gene. The parents of a child with CF each carry one CF gene and one normal gene. They have no symptoms and no disease.
How does CF occur?
When each of the parents contributes a gene to their child, they could pass on either their CF gene or their non-CF gene. Each pregnancy could result in one of three outcomes:
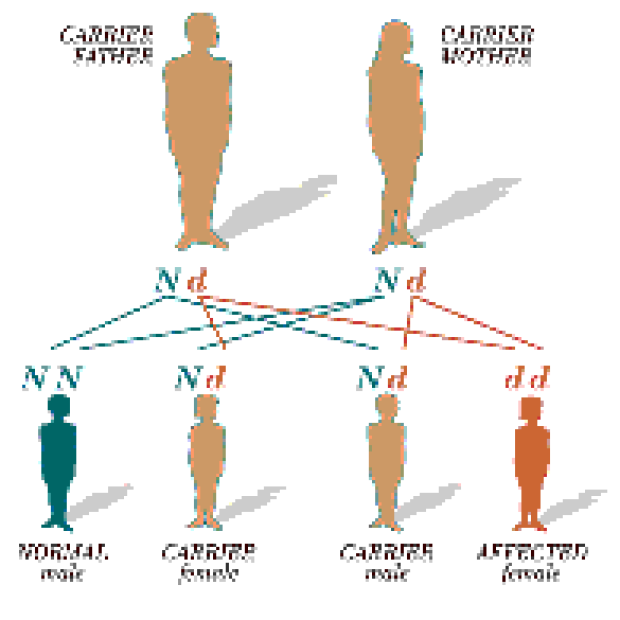
- A one in four (25%) chance that the child will have CF
- A two in four (50%) chance that the child will be a carrier
- A one in four (25%) chance that the child will not carry the CF gene
Can I find out if I have a CF gene?
At the present time, carrier testing is available through a DNA test. If a family member has CF and the gene mutation is known, discovery of the CF gene in other family members can be made with great accuracy. If the specific mutation is not known, the test will be done on the 70%-90% of the CF genes that are most commonly found, but the test won't be 100% accurate. The screening test for people without a family history of CF will also be done on the most common gene mutations, and so cannot be said to be 100% accurate.
CF and the Lungs
What happens in the lungs:
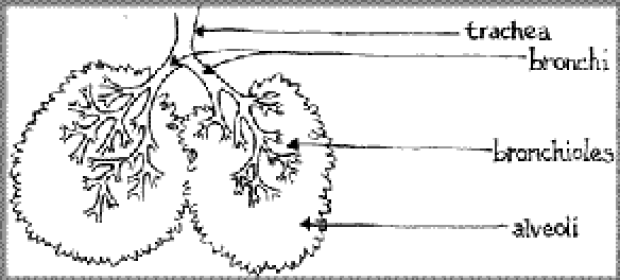
The lungs are like an upside down tree: the trachea is the trunk, the bronchi are the main branches, the bronchioles are smaller branches, and the alveoli are the smallest little twigs and leaves. Normally, tiny hair-like structures known as cilia remove mucus and other substances from the lungs, and bacteria are cleared out. But, because CF produces thick, sticky mucus, the cilia cannot sweep the lungs, and the bacteria remain. This brings about an immunological response from the body's white blood cells: they race to the scene, fight with bacteria, and promptly die
leaving their remains which also contribute to the stickiness of the mucus. Mucus then builds up in the lungs, and lung function starts to drop. It is this residual infection and poor lung functioning that can cause permanent lung damage over time.
What treatment can be done:
The basic daily care program varies to suit individual needs. These are some common pulmonary therapy treatments:
- First an inhaled medication to open up the lung passages
- Then an airway clearance technique to mobilize the thick mucus from the lungs
- Finally medications, if prescribed, to treat infection or help thin mucous
Common Airway Clearance Techniques:
- Chest Physical Therapy: Using cupped hands to clap on the back and chest
- Percussor: A hand-held device that assists in the mobilization of bronchial secretions by manually/mechanically "clapping" the chest wall.
- Flutter: A pocket device that provides positive expiratory pressure (PEP) therapy. It looks like a fat pipe. Inside the pipe is a plastic cone cradling a steel ball sealed with a perforated cover. Exhaling through your mouth into the flutter with a moderate force causes the ball to oscillate (move back and forth) in the pipe. Oscillation is transmitted throughout the airways, loosening secretions.The force of exhalation helps to mobilize secretions.
- Therapy Vest: Known as high frequency chest compression (HFCC), this device is worn like a vest. It works in two ways: the chest wall is vibrated to break up sputum, then chest wall oscillation causes outward airflow, like a miniature cough.
- Autogenic drainage uses the patient's own airflow to mobilize secretions, through controlled, graduated inspiratory and expiratory maneuvers. This technique, though sometimes difficult to learn and do correctly, does not require any assistive devices.
- Active Cycle of Breathing (ACB) Technique is another alternative form of CPT which requires no percussion. This method requires training with a respiratory therapist to perform properly. ACB is combined with a forced expiratory technique (which uses "huffing" from various lung volumes to assist in removal of secretions) and thoracic expansion exercises.
- Intrapulmonary Percussive Ventilation (IPV) is an airway clearance technique that uses compressed gas to deliver a series of pressurized gas minibursts to the respiratory tract usually by a mouthpiece. The IPV device is a pressurized aerosol machine that delivers aerosolized medications through a mouthpiece under pressure and with oscillations that vibrate the chest and loosen airway secretions.
Lung Infections
What Is an Infection?
An infection occurs when pathogenic microorganisms (like bacteria, viruses, or fungi) invade tissues where they don't belong.

What happens in an infection?
- Invaded by these unfriendly organisms, the tissue becomes inflamed, the normal reaction of tissue to injury. Inflammation is characterized by heat, swelling, redness, and pain.
- The invading microorganisms damage lung tissue. Damaged cells send out chemical messages to the body, called chemotactic substances. These chemical messages initiate the body's immune response.
- The immune response attempts to eliminate the invading microorganisms by increasing blood flow to the infected tissues and by mobilizing specialized white blood cells (phagocytes & lymphocytes) to travel to the infected area and attack the invaders.
- As in any battle, many organisms die. These dead cells can accumulate in the lungs in the form of increased mucus.
Why Do People with CF have to worry about all this?
People with CF have thick mucus which can trap microorganisms in the lungs. Thick mucus is hard to remove, so the microorganisms remain in the lungs, growing and reproducing. Once these organisms are established in the lungs, there are more frequent lung infections. Infection weakens the body and the immune system. Repeated infections initiate a cycle of inflammation and infection which soon becomes a chronic condition.
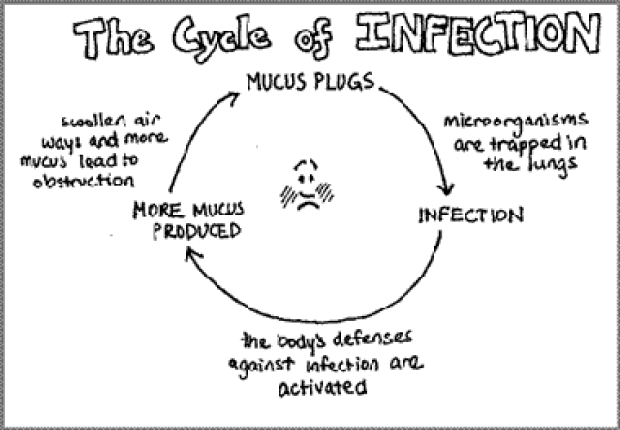
What are the consequences of chronic lung inflammations and infection?
- Tissue damage: microorganisms damage tissue during their invasion, but some of the white blood cells can also damage lung tissue as they attempt to destroy the invading microorganisms.
- Thickened mucus: dead cells can accumulate in the lungs, adding to and thickening the mucus and making it harder to remove.
- Swelling: inflammation of the airways tissue causes swelling which decreases the size of the passageways, making it more difficult to clear mucus.
- Fibrosis: after repeated damage to the lungs, connective tissue forms around the airways this is fibrosis this process can decrease lung elasticity and reduce lung function.
CF and the Digestive System
What is the digestive system?
The mouth is the start of the digestive system. Saliva starts the digestive process. Food is chewed, swallowed, passes down the esophagus into the stomach where more digestion occurs. But, most digestion occurs in the small intestine. It is here that enzymes help break down food so that it can be absorbed into the bloodstream and used for energy. Enzymes are secreted by the pancreas, a small gland located just above the small intestine. The main job of the pancreas is to secrete these enzymes; it also is the place where insulin is made. After enzymes have broken the food down it is absorbed. Any food not broken down passes into the large intestine and is excreted.
What happens in CF?
Mucous plugs can block the pancreas and prevent enzymes from entering the small intestine, which leads to improper digestion of foods. Without these digestive juices, the intestines cannot absorb fats and proteins completely, so nutrients pass out of the body unused. The stools become very large, lighter in color, greasy, and will float on top of the water in the toilet. Unabsorbed fats may also cause excessive intestinal "gas," an abnormally swollen belly, and abdominal pain or discomfort. Weight loss or difficulty maintaining adequate weight can occur. Not all people with CF are pancreatic insufficient.
What can be done?
In general, the same wholesome foods you would give anyone, child or adult, are suitable for the person with CF. The difference will arise in the quantities that are required and the supplements (vitamins and enzymes) needed to ensure sufficient calories. Sometimes there is a need to add salt to replace what is lost in sweat.
Vitamins: Because of the incomplete digestion of fats, fat-soluble vitamins (A, D, E, and K) may be poorly absorbed. Water-soluble supplements are recommended, with the dose varying according to age.
Enzymes: People with CF can take doses of pancreatic enzymes by mouth to help them digest foods better. Pancreatic enzymes help the body absorb nutrients from food, and reduce both the number and bulk of stools, and the amount of flatulence, abdominal pain, and distension.
Minerals: Since large amounts of sodium and chloride are lost in sweat, salt and salty foods are recommended for all ages. Salt tablets, however, aren't usually prescribed.
Oral supplements: Even with enzyme supplements, people with CF absorb food less efficiently. Some may use oral supplements to augment their total caloric intake.
Glossary of Terms
A
Airway Clearance
The removal of mucus secretions from the lungs by coughing or other methods.
Anti-inflammatory
Something that stops swelling
Antibiotics
Medicines that kill bacteria (not viruses)
B
Bronchodilator
Medicine that opens and relaxes the lungs to aid breathing.
C
Carrier
Someone that has one CF gene instead of two. They do not have CF but can give it to their child. For a child with CF, each parent either has CF (two CF genes) or is a carrier (one CF gene).
Chest clearance therapies
Treatments to clear lung mucus (chest physiotherapy, the Vest™, the Flutter®, Acapella™, etc.).
Chest physiotherapy (CPT)
Treatment to break up and loosen lung mucus so that it can be coughed out.
Chronic
Lasting a long time. CF is a chronic disease. The opposite of "chronic" is "acute".
D
Ducts
Tubes or pathways for secretions. Ducts are found in organs, organ systems, and glands. In CF, thick mucus can clog ducts and block secretions.
E
Enzymes
Enymes help to break down foods during digestion. In CF, mucus can block the tube that carries enzymes from the pancreas to the food. People with CF may take extra enzymes to help digest their food.
G
Gastrointestinal
Relating to the stomach and intestines
Gene
The basic unit of heredity. Genes decide a big part of what people are like (eye color, looks, height, health). CF is caused by a defect of a gene. If the Dad's sperm has a CF gene and the Mom's egg has a CF gene, the child will have CF.
Genetic
Having to do with genes (See "Gene"). A trait passed on from one family member to another.
Germs
Viruses or bacteria
Glands
A cell, group of cells, or organ that makes a secretion for use in the body
H
Hormone
A secretion of certain glands. Hormones manage body functions like growth, maturing, and heart rate. Hormones are not affected by CF.
I
Immunizations
Shots needed to protect from illness
Infection control
Stopping the spread of illness by washing, cleaning, avoiding sick people, etc.
Inflammation
The swelling of body tissues due to irritation or injury. Inflammation is a process by which the body's white blood cells and chemicals protect us from infection and foreign substances such as bacteria and viruses. Inflammation occurs with an infection.
Intestinal blockage
Something that blocks the flow of food or feces in the intestines
M
Malabsorption
Poor uptake of nutrients from food for use by the body. In CF, mucus can plugs the ducts that carry the enzymes and hormones used in digestion. The body can't digest food as well so doesn't get the nutrients from the food. The body needs nutrients for health and growth. A common symptom of CF is failure to thrive.
Median
The middle point in a line of values. Above and below the median are an equal number of values. In "1 4 5 9 12", "5" is the median. Two numbers are above the 5, and two numbers are below it.
Mucus
A thin, slippery fluid made by mucus membranes and glands. In CF, mucus is often thick and sticky.
N
Nutrition supplements
Pills, fluids, snacks, and drinks that give the body extra nutrition.
P
Pancreas
Long gland-like organ found behind the stomach. The duct part of the pancreas secretes enzymes into the intestine to help break down food. In CF, mucus may clog the ducts and block digestion. The other part of the pancreas contains endocrine tissue, which makes the hormone insulin. Insulin controls how the body uses and stores sugar.
Pancreatic duct
See "Pancreas"
Pancreatic Enzyme Supplements
See "Enzymes"
S
Secretions
See "Mucus"