Pompe Disease
A type of Lysosomal Storage Disease
What is Pompe disease?
Overview
Pompe disease, (also known as Glycogen Storage Disease type II), is a type of lysosomal storage disease, meaning there is an issue in how parts of the cell (lysosomes) break down certain molecules. Pompe disease is a genetic condition that may be detected by Newborn Screening or may be diagnosed at various ages due to different symptoms. There are two major classifications of Pompe disease.
- 1. Classic Infantile-Onset Pompe Disease- symptoms occur before 12 months and usually include heart muscle thickening (cardiomyopathy), muscle weakness, low tone, feeding difficulties, and respiratory distress
- 2. Late-Onset Pompe Disease- symptoms occur before 12 months without heart muscle thickening (cardiomyopathy), Symptoms occur after 12 months with muscle weakness and respiratory difficulties
Cause
Pompe disease is a genetic condition. This means that it is caused by a defect in a gene, or a specific instruction the body uses to function normally. The specific instruction that is impaired in Pompe disease is a gene called GAA. The GAA gene has instructions to make an enzyme called acid alpha-glucosidase. This enzyme is important in breaking down stored sugar in the body (glycogen). In Pompe disease, this enzyme activity is low, leading to glycogen accumulation in multiple organs throughout the body.
To have this condition a person must have inherited two abnormal copies of the GAA gene, one from the egg and one from the sperm. This is known as a automal recessive condition.
Surveillance for Pompe disease
When newborn screening is positive for Pompe disease, regular follow-up is needed for your child. You will be referred to our hospital for confirmatory testing. Your child will likely see medical genetics and neurology and may also see cardiology and physical therapy. Different care approaches will be used depending on the type of Pompe disease your child has and your child's unique case.
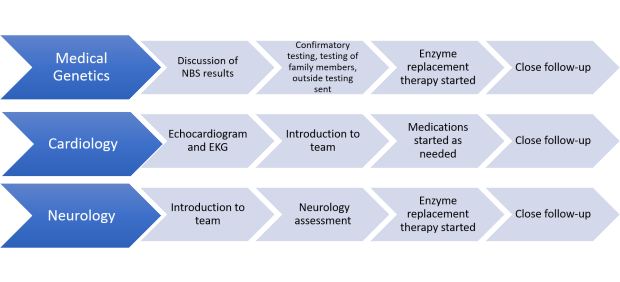
Classic Infantile-Onset Pompe Disease
After a positive newborn screen, the following steps will occur:
•Confirmatory testing
•Testing family members
•If diagnosis is confirmed, it will be discussed with the family and multiple specialists
•Treatment will be started as soon as possible
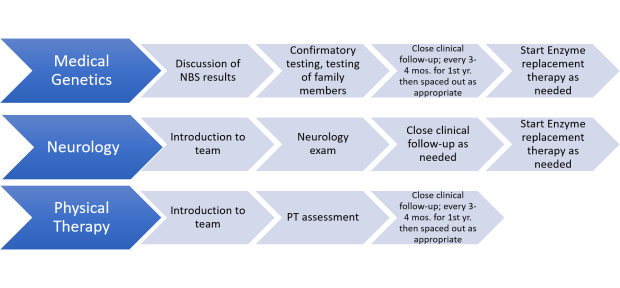
Late Onset Pompe Disease
After a positive newborn screen, the following steps will occur:
•Confirmatory testing
•Testing family members
•If diagnosis is confirmed, your child will be follow closely, especially in the first year of life by multiple specialists
•Treatment will be started as needed
Treatment & Research for Pompe Disease
Treatment for Pompe disease are considered case by case, and research options may also be available. Options are listed below:
- Enzyme replacement therapy
- Active research with Dr. John Day’s group: http://med.stanford.edu/day-lab/clinical_trials.html
- Consult clinicaltrials.gov
Mucopolysaccharidosis type I
A type of Lysosomal Storage Disease
What is Mucopolysaccharidosis type I (MPSI)?
Overview
Mucopolysaccharidosis type I (MPSI) is type of lysosomal storage disorder, meaning there is an issue with how parts of a cell (lysosomes) break down certain molecules. MPSI is a genetic condition that may be diagnosed based on a variety of symptoms. Symptoms range in severity and age of onset. The two main clinical presentations are:
- 1. Severe (Hurler syndrome)- marked by progressive abnormalities in multiple organs, especially the brain, skeleton, and connective tissue; appears in first year of life
- 2. Attenuated (Hurler-Scheie and Scheie syndromes)- a milder version of the disease affecting primarily the joints and skeleton but also affecting also affecting other organ systems; appears in childhood
The state of California plans to start screening newborns for MPSI and Stanford is an area service center. The newborn screen will measure IDUA enzyme activity from blood spots. A positive newborn screen will not be diagnostic. Newborns with low IDUA activity in blood spots on two different occasions will be evaluated by our geneticists and additional tests are needed to confirm the diagnosis.
Cause
As mentioned, MPSI is genetic condition. This means it is caused by a defect in a gene, or a specific instruction the body uses to function normally. The specific instruction that is impaired in MPSI is a gene called IDUA gene. This gene makes an enzyme called IDUA, that allows the body to properly break down certain molecules in our body, called glycosaminoglycans. Without appropiate function of this enzyme, glycosaminoglycans accumulate in different tissues.
To have this condition a person must have inherited two abnormal copies of the IDUA gene, one from the egg and one from the sperm. This is known as a automal recessive condition.
Treatment & Research for MPSI
Appropriate treatment is established based on the level of enzyme activity and the symptoms.
- Patients with attenuated MPSI receive enzyme replacement therapy (link to ERT service)
- Patient with severe MPSI are candidates for hematopoietic stem cell transplantation
Dr. Natalia Gomez-Ospina is a physician scientist whose research interests are in developing a safer, and more effective treatment for MPSI.
The patient’s blood stem cells can be genetically modified to secrete high levels of the missing enzyme and transplanted back into patient where the enzyme secreting cells will repopulate the blood and migrate to affected organs. This is a one-time treatment which circumvents many of the caveats of hematopoietic stem cell transplants and enzyme replacement therapy. It could reduce the time to intervention and decrease complications by eliminating the risk of graft-versus-host disease and need for immunosuppression.
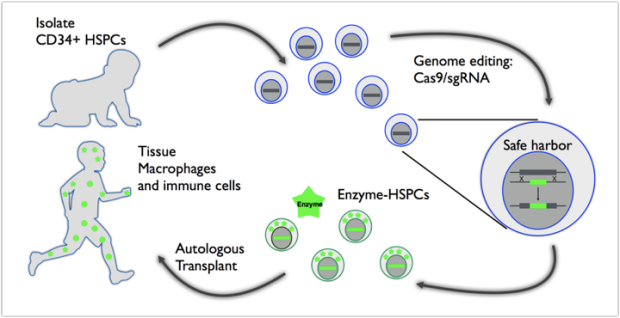
A number of additional therapies may be available in the future. Patients/families may also consider participation in research studies, that may be found by visiting: http://clinicaltrials.gov
Your Stanford Care Team
Medical Genetics
- Greg Enns, MD
- Chung Lee, MD
- Natalia Gomez-Opsina, MD, PhD
- Dena Matalon, MD
Pediatric Stem Cell Transplantation
- Ami Shah, MD
