Basic Research
Metabolism Drives Cell Fate Decisions During Hematopoiesis
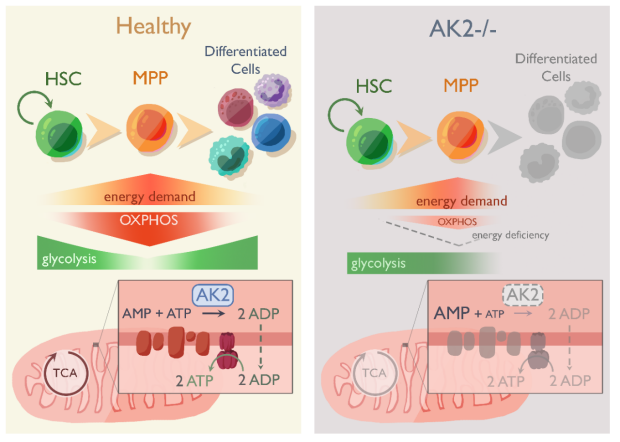
It is an emerging paradigm that changes in metabolism play an intricate role during development and differentiation. In turn, perturbations in metabolism can arrest proliferation and lead to a complete standstill in maturation. We use a mono-genetic disease with defect in oxidative phosphorylation to understand the role of metabolism during the development of different hematopoietic lineages.
Reticular Dysgenesis (RD) is one of the most serious forms of severe combined immunodeficiency (SCID) because it affects both, the innate and adaptive immune system. In addition to lymphopenia, typical for classic SCID, RD patients also have severe congenital neutropenia and sensorineural hearing loss. Invariably, RD patients die early in life unless immune reconstitution is achieved by hematopoietic stem cell transplantation (HSCT). RD is caused by biallelic mutations in the mitochondrial enzyme Adenylate Kinase 2 (AK2) which catalyzes the phosphorylation of adenosine monophosphate (AMP) to adenosine diphosphate (ADP) for oxidative phosphorylation. Unlike other forms of SCID, RD is a mitochondrial disease. How this mitochondrial defect causes a maturation arrest across the myeloid and lymphoid lineages, has never been elucidated.
We have developed an elegant biallelic CRISPR-model of RD in primary hematopoietic stem cells to query the metabolic vulnerability during development. Intriguingly, this system revealed profound and unexpected biochemical stressors…
The Thymus -
An Essential Immune Organ With Untapped Therapeutic Potential
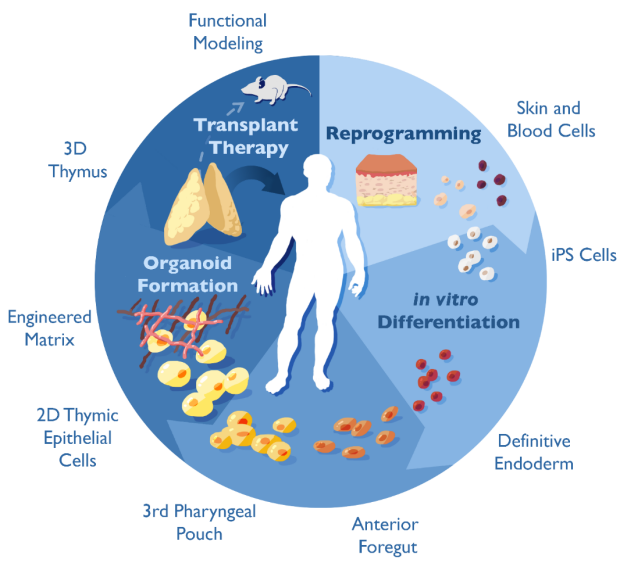
The Thymus Regeneration Project: The thymus is a central immune organ in which specialized thymic epithelial cells direct T cell maturation, specificity and function. Through positive and negative selection, the thymus instructs T cells to recognize vital self from injurious non-self, ultimately giving rise to a diverse and self-tolerant T cell repertoire that protects from infection and cancer and prevents autoimmunity. The thymus peaks in size in infancy and structurally disappears as we age without capacity to regenerate. This process is responsible for the decline in immune competence with age (immune senescence). Due to its enigmatic nature, the thymus’ extraordinary potential to instruct immune function for therapeutic purposes has not yet been tapped into. Our goal is to create transplantation-compatible, functional thymic epithelial organoids by differentiating induced pluripotent stem cells (iPSCs) in vitro into thymic epithelial cells embedded within an engineered extracellular matrix (eECM). Functional thymic epithelial organoids could be used across a wide range of clinical applications to reconstitute and modulate T cell function.
22q11 Deletion Syndrome (22q11 DS) is one of the most common human microdeletion syndromes. Thymic aplasia, often referred to as complete DiGeorge Syndrome, was recognized early on as one of the classic features of 22q11 DS. The prognosis and treatment of thymic aplasia, however, has not significantly changed over the past 20 years. From Dr. Louise Markert’s pioneering clinical studies using allogeneic thymic tissues to treat patients with congenital absence of the thymus, we have learned that donor-derived thymic epithelial cells (TECs) can instruct the development of a diverse T-cell receptor repertoire in the recipient. Lack of graft availability and graft histocompatibility, however, have limited the widespread use of this lifesaving procedure. Making functional, HLA-compatible TECs derived from iPSCs in vitro could overcome previously encountered limitations and holds promise as alternative tissue source for regenerative cell therapies in the future. Using iPSCs from patients with congenial thymic defects as a platform for disease modeling allows us to study the molecular mechanisms underlying thymic abnormalities during ontogeny.
Illustrations by Lara Ellen Bouchard