Publications
Integrated Trajectories of the Maternal Metabolome, Proteome, and Immunome Predict Labor Onset
Science Translational Medicine 05 May 2021:
Vol. 13, Issue 592, eabd9898
DOI: 10.1126/scitranslmed.abd9898
Existing methods for predicting the day of labor perform poorly. In current clinical practice, the estimate day of delivery (EDD) is calculated on the basis of the first day of the last menstrual period (LMP) and assumes a gestational length of 40 weeks. The gestational age (GA) and EDD are further determined by the first accurate ultrasound examination. Although they are useful for managing the pregnancy, the EDD and GA are not accurate predictors for when labor will actually occur because most pregnancies deviate from the norm of 40 weeks of gestational duration. To advance clinical decision-making, further estimation approaches including predictive biomarkers are critically needed to better predict the actual labor onset, leading to delivery in healthy and pathological pregnancies.
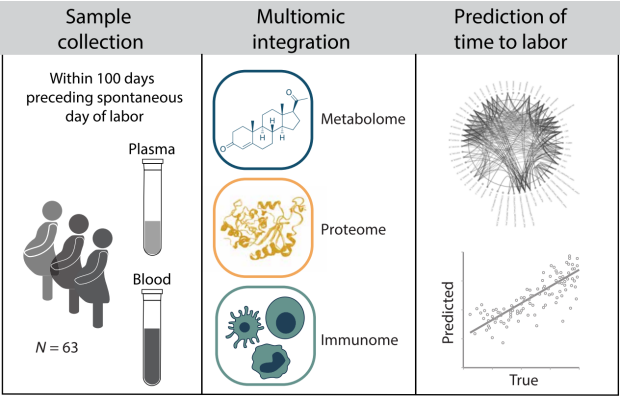
In this study, we combined an untargeted mass spectrometry approach with an aptamer-based technology to quantify the concentrations of 4846 metabolomic and proteomic analytes in longitudinally collected plasma samples during the 100-day period preceding spontaneous labor onset. In parallel, we used a single-cell mass cytometry immunoassay to quantify the dynamic changes in the distribution and intracellular signaling responses of all major innate and adaptive peripheral immune cells (2296 features). The analysis generated three high-dimensional omic datasets. We applied a stacked generalization (SG) algorithm to the multiomic dataset to build and independently validate an integrated model that predicted the time to labor (TL). Model component trajectories revealed precisely timed alterations that marked a transition from pregnancy maintenance to prelabor biology. Our findings and predictive modeling approach can serve to identify elements of a common pathway that precedes labor in term as well as pre- or postterm pregnancies.
Multiomics Characterization of Preterm Birth in Low- and Middle-Income Countries
Alliance for Maternal and Newborn Health Improvement, the Global Alliance to Prevent Prematurity and Stillbirth, and the Prematurity Research Center at Stanford University
JAMA Netw Open. 2020;3(12):e2029655.
doi:10.1001/jamanetworkopen.2020.29655
Preterm birth (PTB) is defined by the World Health Organization as the delivery of a live infant before the completion of 37 weeks of gestation. The worldwide rate of PTB in 2014 was estimated to be 10.6% (uncertainty interval, 9.0%-12.0%), with 80% of all cases occurring in South Asia and sub-Saharan Africa. Many risk factors for PTB have been highlighted in previous studies and include obstetrical (eg, previous PTB and multiple gestation), medical (eg, maternal obesity, diabetes, and chronodisruption), and external (eg, smoking and maternal stress) conditions. For example, a meta-analysis of individual- and population-level attributes among 4.1 million births concluded that “unknown factors requiring further research to act upon account for ~2/3 of the preterm birth rate.” Unveiling and elucidating the role of early biological antecedents of PTB has been deemed a necessary step toward developing new diagnostic tests and therapeutic interventions. Biological investigations into the mechanisms of PTB are complicated, as indicated by accumulating evidence that distinct patient subpopulations follow divergent biological trajectories. Given this heterogeneity, simultaneously studying diverse cohorts is critical for identification of generalizable biological pathways.
Recent technological advances have enabled the characterization of a broad range of biological changes during pregnancy. Biological layers explored include single-cell profiling of signaling pathways, measurements of plasma cell-free ribonucleic acid (cfRNA), proteome and metabolome characterization of the microbiome, and detailed genomics analysis. In addition, a recent multiomics investigation demonstrated that biological changes during normal pregnancy involve a number of intricate interactions of biological processes, which can be measured using a coordinated set of assays. The integration of the large, multidimensional data sets generated in a multiomics setting requires complex machine learning pipelines that will remain robust in the face of the inconsistent intrinsic properties of these high-throughput assays and cohort-specific variations.
To our knowledge, this is the first multiomics analysis of term and preterm pregnancies from multiple cohorts in low- and middle-income countries (LMICs). These cohorts were established using biorepositories of samples and phenotypic data for studying maternal and fetal outcomes collected and stored from diverse populations of South Asia and sub-Saharan Africa. The study aimed to investigate the ability of transcriptomics and proteomics profiling of blood plasma and metabolomics analysis of urine to identify early biological measurements associated with PTB.
Towards personalized medicine in maternal and child health: integrating biologic and social determinants
In collaboration with Stanford Prematurity Research Center (David K. Stevenson, MD)
Pediatric Research
Volume 89, pages252–258 (2021)
Published: 26 May 2020
What is unique about personalized (or rational) medicine in maternal and child health is that it is fundamental to the life course approach to precision health—to predict, prevent, and cure precisely.70 The fetal origin of adult disease is now a popular notion, and there is little doubt that prenatal influences contribute to postnatal conditions that emerge throughout the lifespan. Moreover, both paternal and maternal health are relevant to pregnancy outcomes, and the life course of the fetus certainly begins at conception in ways which are not fully understood. We have suggested before that attention to pregnancy is in our world’s best interest even though the return on investment would likely occur later in the life course of both the mother and the child.
Indeed, political gratification might be delayed, but savings in healthcare costs would ultimately be realized. For example, the increased risk of heart disease in women after preeclampsia71 could be alleviated or PTB could be decreased on a large scale by decreasing the occurrence of preeclampsia, or at least if babies could be born in a healthier disposition later in gestation, closer to term.
Metabolic Dynamics and Prediction of Gestational Age and Time to Delivery in Pregnant Women
From the Stanford laboratory of Michael Snyder, Ph.D
Cell Press
Volume 181, Pages 1680–1692
June 25, 2020
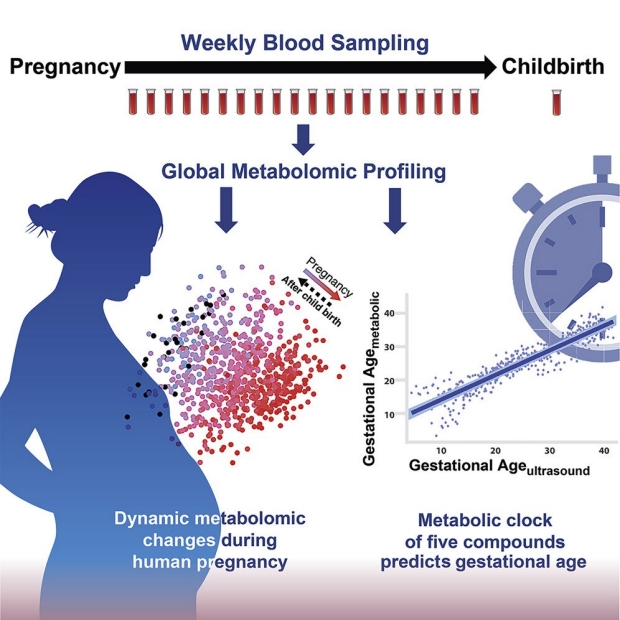
The maternal circulatory system connects with the fetal circulatory system through the placenta, carrying bioactive molecules and biomarkers such as steroid hormones, micronutrients, and circulating nucleic acids, whose concentrations alter as gestation progresses. Recent work on cell-free RNA suggests that markers in maternal blood can be used to estimate gestational age, but sequencing can be expensive and time-consuming, and the accuracy, at present, is not ideal. Therefore, a more accurate and cost-effective method for estimating gestational age and delivery time, possibly using blood metabolites, is needed. In addition, current clinical tests often only focus on a few markers, whereas research covering more molecules often examines the profiles at one or a few time points during pregnancy. Thus, a high-resolution landscape of pregnancy-related metabolites during healthy pregnancy and the postpartum period is still poorly understood.
Here, we use untargeted metabolomics to systematically profile blood metabolites throughout pregnancy with weekly sampling of maternal blood. The study identified a large number of pregnancy-related metabolites and metabolic pathways offering a comprehensive view of the metabolite changes during healthy pregnancy and the postpartum period. Leveraging the high-resolution datasets, we built a metabolic clock that not only predicts gestational age in high accordance with the first-trimester ultrasound, the clinical gold standard, but also recovers personal pregnancy variations undetected by ultrasound but capable of affecting delivery time.
Multiomic immune clockworks of pregnancy
In collaboration with Stanford Prematurity Research Center (David K. Stevenson, MD)

Semin Immunopathol
Issue 42, Pages 397–412 (2020)
https://doi.org/10.1007/s00281-019-00772-1
For the establishment, maintenance, and completion of mammalian pregnancy, the maternal immune system must adhere to a precise schedule. During 9 months, dynamic local and systemic immune changes occur that confer tolerance to the semi-allogenic fetus while protecting the mother against invading pathogens. The appropriate execution of these important events requires a tightly regulated immunologic timeline governed by a complex system of immune pacemakers. Severe pregnancy complications, such as preterm labor and preeclampsia, can result when these immunological adaptations are disrupted.
Over six decades of research have contributed to our current understanding of the chronology of feto-maternal immune adaptations during pregnancy, and the mechanisms of maternal immune tolerance to the developing fetus have been extensively reviewed. However, the feto-maternal immune system does not evolve in isolation but rather as a component of a complex network of endocrine, metabolic, and microbiome adaptations that interact with signals from the fetus and the placenta in a timely, coordinated manner. Understanding this immune clock is of paramount importance when addressing the problem of prematurity, as a preponderance of evidence has linked immune dysregulation with not only preterm labor but also diseases of pregnancy such as intrauterine growth restriction and preeclampsia, which are major indications for preterm delivery. An integrated examination of the factors that influence the programming of immune adaptations during gestation is thus essential to advance our knowledge of both healthy and pathological pregnancies.
The recent advent of high-content transcriptomic, epigenomic, proteomic, and cytomic technologies has provided powerful means to capture the complexity of multiomic adaptations during pregnancy. Specifically, a network of interrelated immune features that are chronologically regulated over the course of gestation has recently been demonstrated. In this review, we will focus on the fetal, placental, and maternal pacemakers that program this immune clock of pregnancy and highlight recent technological advances that allow an integrated, multiomic assessment of immunological events involved in the natural chronology of pregnancy.
Multiomics modeling of the immunome, transcriptome, microbiome, proteome and metabolome adaptations during human pregnancy
In collaboration with Stanford Prematurity Research Center (David K. Stevenson, MD)
Mohammad Sajjad Ghaemi, Daniel B DiGiulio, Kévin Contrepois, Benjamin Callahan, Thuy T M Ngo, et al.
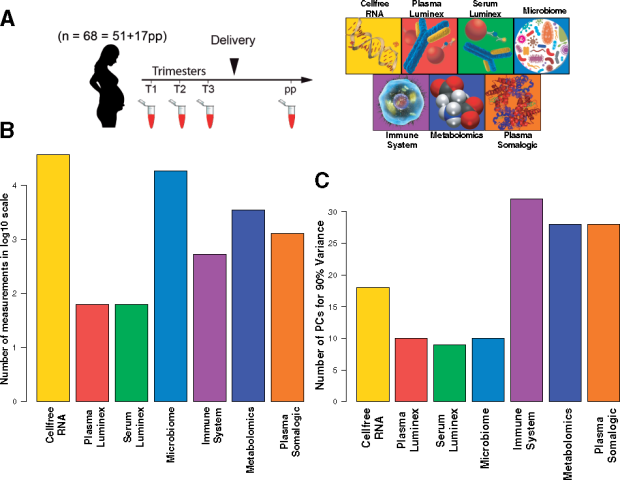
Bioinformatics
Volume 35, Issue 1, Pages 95–103 (2019)
https://doi.org/10.1093/bioinformatics/bty537
Physiological changes during pregnancy are highly dynamic and involve coordinated changes among multiple interconnected molecular and cellular systems from the fetus, the fetal-membrane and the mother. The simultaneous interrogation of these systems can reveal otherwise unrecognized crosstalk. Understanding such crosstalk can inform several lines of investigation. From a biological perspective, it can point to important disease mechanisms such as immune programming by the microbiome, or specific interactions between proteins and cellular elements . From a diagnostic perspective, it can reveal biomarkers from several biological domains that provide higher predictive power if combined. Alternatively, it can point to alternative biomarkers in an accessible biological compartment, which can replace biomarkers that are difficult to obtain or expensive to measure.
The main objective of this study was to test multiple strategies for integrating transcriptomic, immunological, microbiomic, metabolomic and proteomic datasets into different statistical models predicting gestational age in term pregnancy and identify the most accurate strategy. A final objective was to interrogate the derived model for novel and testable biological hypothesis
Cross-Platform Comparison of Untargeted and Targeted Lipidomics Approaches on Aging Mouse Plasma
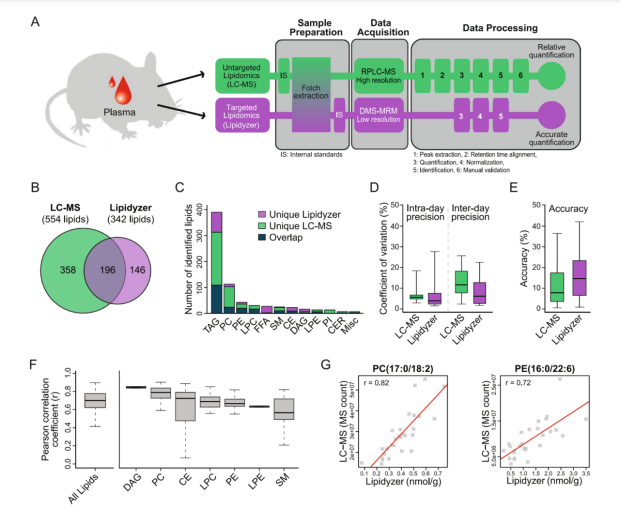
Scientific Reports
Volume 8, Article number: 17747 (2018)
- DOI: 10.1038/s41598-018-35807-4
Lipids play a key role in many biological processes, such as cellular structure, energy storage, and cell signaling, and are dysregulated in a variety of health conditions, including aging, cancer, diabetes, cardiovascular disorders and neurodegenerative diseases. As such, there is a strong interest in the comprehensive analysis of lipids for biomarker discovery and for gaining novel insights into the onset and progression of diseases. However, global profiling of the entire collection of lipids (known as lipidomics) is challenging because the lipidome comprises thousands of different molecular species that are highly diverse in chemical structure and composition.
Mass spectrometry (MS) is the preferred technology to profile lipids as it provides resolution, sensitivity, selectivity, and throughput. MS-based lipidomics can be performed using either untargeted or targeted approaches, each with their own set of advantages and limitations. Untargeted platforms are unbiased and have broad coverage, as they potentially detect all the lipids present in a sample. However, they are often used in a semiquantitative manner (providing relative quantification) and certain aspects of the workflow, including data normalization and lipid identification, are challenging, time-consuming and not well standardized. Untargeted lipidomics can be done by direct infusion or liquid chromatography (LC)-based approaches. LC-MS is often preferred because it offers higher sensitivity as well as more accurate lipid identification and quantification. In contrast, targeted platforms traditionally focus on the absolute quantification of a small number of pre-defined lipids using isotopically labeled internal standards. The number of targets is often limited due to the lack of commercially available standards. Targeted approaches are high-throughput because data generation and analysis is fast and straightforward, quantitative, but usually have a more limited coverage.
Recently, a novel targeted lipid analysis technology has been developed called the Lipidyzer platform (SCIEX) that can quantify over 1,100 lipid molecular species across 10 lipid classes in human plasma/serum. The Lipidyzer platform estimates concentrations using a mixture of internal standards specifically designed for this purpose. Consequently, the Lipidyzer platform may constitute a good compromise between untargeted and conventional targeted approaches, providing broad coverage, accurate quantification and fast and straightforward data processing. However, its performance has not been compared to more established untargeted lipidomics approaches and it has not been tested in a biological setting.
These questions prompted us to compare a conventional untargeted LC-MS approach with the targeted Lipidyzer platform. In this study, we first conducted a cross-platform comparison of the workflow, lipid coverage, precision and accuracy, and determined how well the two platforms correlate quantitatively with each other. We then applied both platforms to profile lipids in plasma from young and old mice, because a comprehensive description of changes in lipids during healthy aging in mice is still lacking. Altogether, our study constitutes the first cross-platform comparison of two state-of-the-art lipidomics platforms and provides the first assessment of the Lipidyzer platform in the context of aging.
Can Metabolic Profiles Be Used as a Phenotypic Readout of the Genome to Enhance Precision Medicine?
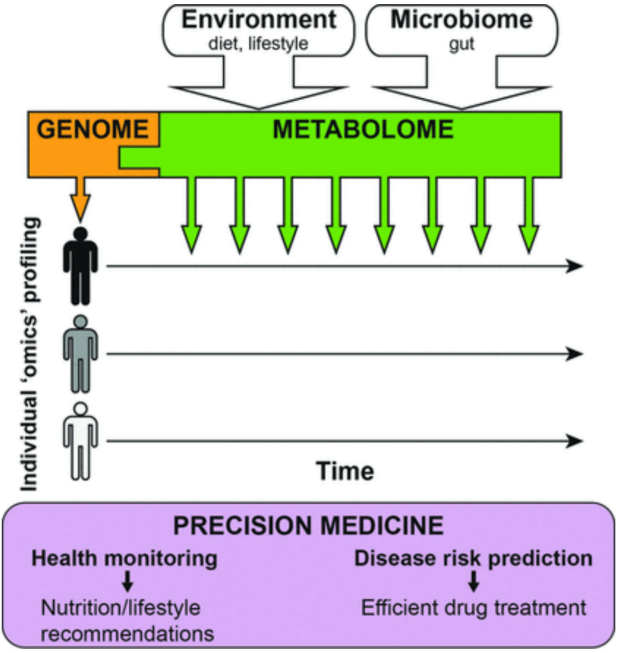
Clinical Chemistry
Volume 62, Issue 5: Pages 676-678 (2016)
DOI: 10.1373/clinchem.2015.251181.
Combination of personal genetic information and metabolic profiles for precision medicine
Static DNA sequences inform on inherited genetic variations, whereas dynamic metabolites—influenced by the environment and the microbiome—give access to the phenotypic readout of the genome. After defining an individual health baseline, regular metabolome measurements (indicated by the multiple green down arrows) allow health monitoring as well as disease risk prediction and can result in nutrition/lifestyle recommendations to prevent the onset of a disease or increase the efficiency of drug treatments.
Optimized Analytical Procedures for the Untargeted Metabolomic Profiling of Human Urine and Plasma by Combining Hydrophilic Interaction (HILIC) and Reverse-Phase Liquid Chromatography (RPLC)-Mass Spectrometry.
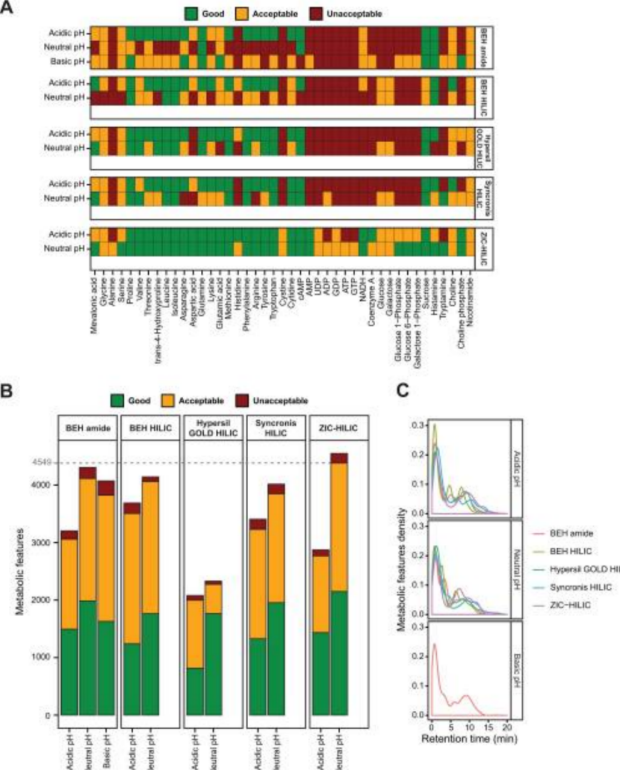
Molecular and Cellular Proteomics.
2015 Jun;14(6):1684-95.
DOI: 10.1074/mcp.M114.046508
Metabolomics is a relatively recent “omic” that aims at measuring the quantity of a large collection of metabolites (i.e. low-molecular-weight organic compounds, typically < 1,500 Da). It is often applied to the study of human diseases (i.e. characterization of deregulated metabolic pathways and discovery of therapeutic targets and biomarkers), drug toxicity and efficacy, and environmental exposure (e.g. food) and lifestyle (e.g. fitness) on health. Metabolomics is advantageous over other “omics” (i.e. genomics, transcriptomics, and proteomics) because it measures a more direct functional readout of activity and phenotype. When applied to biofluids (i.e. urine and blood), the profiling of metabolites reveals a snapshot of the “metabolic status” of the subject and as such holds great promise for personalized metabolomics and medicine .
Metabolic profiling studies are mostly performed using i) chromatography coupled to mass spectrometry (MS) instruments, including gas chromatography (GC)-MS and liquid chromatography (LC)-MS, as well as ii) nuclear magnetic resonance (NMR) spectroscopy platforms. Few studies have highlighted the benefit of combining multiplatform approaches for the analysis of urine and blood. However, due to instrumentation limitation, most laboratories use a single analytical approach. Because of its high sensitivity and wide range of metabolites that can be analyzed, LC-MS utilization has expanded rapidly over the past 10 years. Most untargeted studies are performed using reverse-phase liquid chromatography (RPLC, mainly C18-bonded silica columns) because it generates reproducible data for a large set of metabolites (non- and moderately polar compounds). However, many metabolites in biofluids are water soluble, polar, and ionic (e.g. amino acids, organic acids, sulfates, and sugars) and are usually not retained on RPLC columns, thus hindering their identification and accurate quantification
Biallelic Mutations in ATP5F1D, which Encodes a Subunit of ATP Synthase, Cause a Metabolic Disorder.
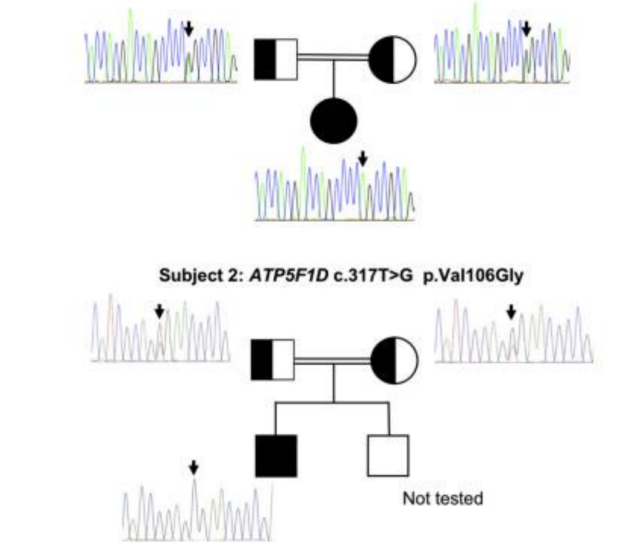
American Journal of Human Genetics
2018 Mar 1;102(3):494-504.
DOI: 10.1016/j.ajhg.2018.01.020.
Mitochondrial diseases are clinically and genetically heterogeneous. Findings such as hyperammonemia, lactic acidosis, and rhabdomyolysis suggest mitochondrial dysfunction and can occur as a result of defects in fatty acid oxidation as well as disorders of the respiratory chain. Defects in the electron transport chain (ETC), which underlies oxidative phosphorylation (OXPHOS), can be caused by mutations in the nuclear or mitochondrial genome. Accordingly, inheritance can be autosomal, sex linked, or maternal. Presentations vary widely and range from lethal neonatal metabolic decompensation to chronic progressive disorders of adulthood.
Complex V is the final multi-subunit complex of the OXPHOS system. It harnesses energy from the proton electrochemical gradient to synthesize ATP from ADP3 and inorganic phosphate, which is the main source of energy for intracellular metabolic pathways.4 Mitochondrial ATP synthase consists of two main functional domains, the soluble F1 catalytic portion in the mitochondrial matrix and the inner-membrane-embedded FO, which allows protons to pass from the intermembrane space to the matrix (reviewed by Jonckheere et al.). Two subunits of the FO (a and A6L) are encoded by mtDNA (MT-ATP6 and MT-ATP8), whereas the other subunits and accessory factors are encoded by the nuclear genome. Although mitochondrial disorders due to defects in mitochondrial complex V have been reported, they are very rare in comparison with those due to mutations in the genes encoding the proteins of the other complexes (I–IV)