LABORATORY RESEARCH
The Ye Lab is currently investigated three areas of cancer metabolism resarch:
Project 1:
a. Deciphering the Warburg effect: mitochondrial uncoupling induces metabolic reprogramming, epigenetic remodeling and cell differentiation.
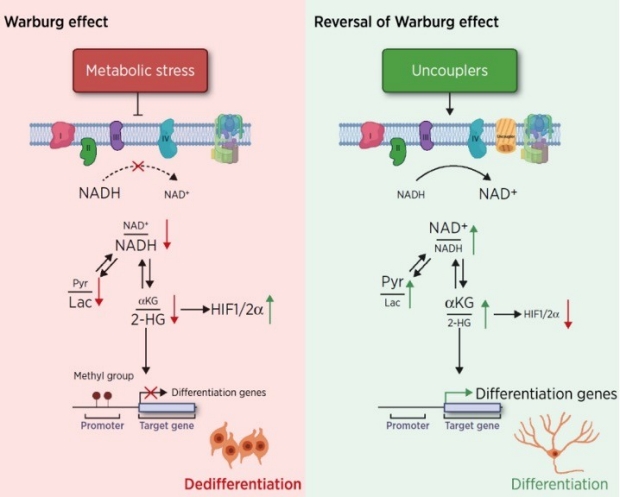
The Warburg effect is the major metabolic hallmark of cancer. According to Warburg himself, the consequence of the Warburg effect is cell dedifferentiation. Therefore, reversing the Warburg effect might be an approach to restore cell differentiation in cancer. In this study, we used a mitochondrial uncoupler, niclosamide ethanolamine (NEN), to activate mitochondrial respiration, which induced neural differentiation in neuroblastoma cells (Jiang et al., Cancer Research, 2023).
- NEN treatment increased the NAD+/NADH and pyruvate/lactate ratios and also the α-ketoglutarate/2- hydroxyglutarate (2-HG) ratio.
- NEN treatment induced promoter CpG island demethylation and epigenetic landscape remodeling, activating the neural differentiation program.
- NEN treatment upregulated p53 but downregulated N-Myc and β-catenin signaling in neuroblastoma cells.
- Even under hypoxia, NEN treatment remained effective in inhibiting 2-HG generation and suppressing hypoxia-inducible factor (HIF) signaling.
- Dietary NEN intervention reduced tumor growth rate, 2-HG levels, and expression of N-Myc and β-catenin in tumors in an orthotopic neuroblastoma mouse model.
- Integrative analysis indicated that NEN treatment upregulated favorable prognosis genes and downregulated unfavorable prognosis genes.
Altogether, these results suggest that mitochondrial uncoupling is an effective metabolic and epigenetic therapy for reversing the Warburg effect and inducing differentiation in neuroblastoma.
b. Mitochondrial uncoupling inhibits reductive carboxylation in cancer
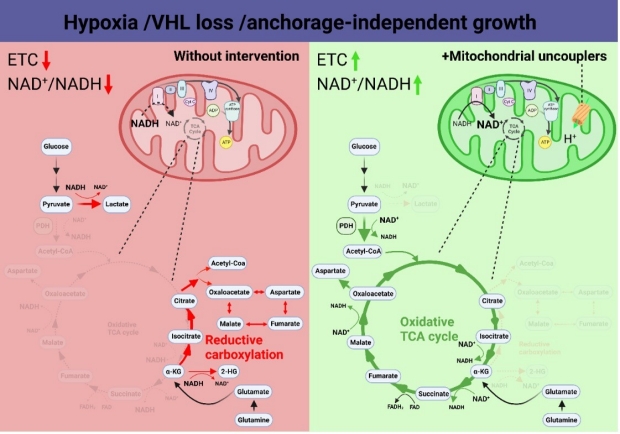
When the electron transport chain (ETC) function is impaired, cancer cells rely on reductive carboxylation (RC) to convert α-ketoglutarate (αKG) to citrate for macromolecular synthesis, thereby promoting tumor growth. Currently, there is no viable therapy to inhibit RC for cancer treatment. Recently we demonstrated that mitochondrial uncoupling accelerates the oxidative tricarboxylic acid (TCA) cycle and blocks RC under hypoxia, in von Hippel-Lindau (VHL) tumor suppressor–deficient kidney cancer cells, or under anchorage-independent growth condition. Together, these data demonstrate that mitochondrial uncoupling redirects α-KG from RC back to the oxidative TCA cycle, highlighting that the NAD+/NADH ratio is one key switch that determines the metabolic fate of α-KG (Jiang et al., Mol Cancer Res. 2023).
Future directions:
1. Assess the therapeutic efficacy of mitochondrial uncouplers across diverse cancer types.
2. Explore the synergistic effects of combining mitochondrial uncouplers with other therapies for cancer treatment.
3. Elucidate the mechanisms by which mitochondrial uncouplers induce epigenetic remodeling.
Project 2: Deciphering the Warburg effect: diverting pyruvate flux away from TCA cycle to reduce histone acetylation and chromatin accessibility.
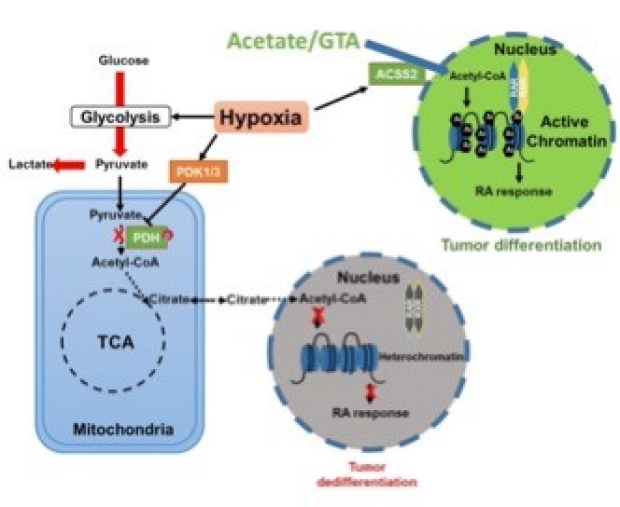
Despite the fact that Otto H. Warburg discovered the Warburg effect almost one hundred years ago, why cancer cells waste most of the glucose carbon as lactate remains an enigma. Warburg proposed a connection between the Warburg effect and cell dedifferentiation. Hypoxia is a common tumor microenvironmental stress that induces the Warburg effect and blocks tumor cell differentiation. Using a neuroblastoma differentiation model, we discovered that:
- Hypoxia repressed cell differentiation through reducing cellular acetyl-CoA levels, leading to reduction of global histone acetylation and chromatin accessibility.
- The metabolic switch triggering this global histone hypoacetylation was the induction of pyruvate dehydrogenase kinases (PDK1 and PDK3). Inhibition of PDKs using dichloroacetate (DCA) restored acetyl-CoA generation and histone acetylation under hypoxia.
- Knocking down PDK1 induced neuroblastoma cell differentiation, highlighting the critical role of PDK1 in cell fate control.
- Importantly, acetate or glycerol triacetate (GTA) supplementation restored differentiation markers expression and neuron differentiation under hypoxia.
- ATAC-Seq analysis demonstrated that hypoxia treatment significantly reduced chromatin accessibility at RAR/RXR binding sites, which can be restored by acetate supplementation.
Together, these findings suggest that diverting pyruvate flux away from acetyl-CoA generation to lactate production is the key mechanism that Warburg effect drives dedifferentiation and tumorigenesis. We propose that combining differentiation therapy with acetate/GTA supplementation might represent an effective therapy against neuroblastoma (Li et al., Cell Death & Dis 2020).
Future directions:
1. Clarify the regulatory mechanisms governing the pyruvate dehydrogenase complex at both transcriptional and post-translational levels.
2. Examine the impact of acetyl-CoA metabolism compartmentalization on histone acetylation and transcriptional regulation.
3. Explore the therapeutic efficacy of PDK inhibitors in promoting tumor differentiation.
Project 3: Serine and one-carbon unit metabolism in epigenetic regulation and breast cancer progression

a. Metabolic Profiling Reveals a Dependency of Human Metastatic Breast Cancer on Mitochondrial Serine and One-Carbon Unit Metabolism
Metastasis is the leading cause of death in breast cancer. We performed metabolomics profiling on the triple negative breast cancer cell line MDA-MB-231 and its metastatic subclones to determine whether metastatic breast cancer cells develop new metabolic properties. Here, we report that these metastatic subclones display an altered metabolic profile compared to the parental population.
- The mitochondrial serine and one-carbon (1C) unit pathway is upregulated in metastatic subclones.
- Inhibition of the first rate-limiting enzyme of the mitochondrial serine and 1C unit pathway, serine hydroxymethyltransferase (SHMT2), suppresses metastatic subclone proliferation in culture and impairs growth at both primary and metastatic sites in mice.
- Some human breast cancers exhibit a significant association between the expression of genes in the mitochondrial serine and 1C unit pathway with disease outcome and higher expression of SHMT2 in metastatic tumor tissue compared to primary tumors.
Together, these results reveal an important role for mitochondrial serine and 1C unit metabolism in supporting metastatic growth in a subset of human breast cancers (Li et al., Mol Cancer Res. 2020).
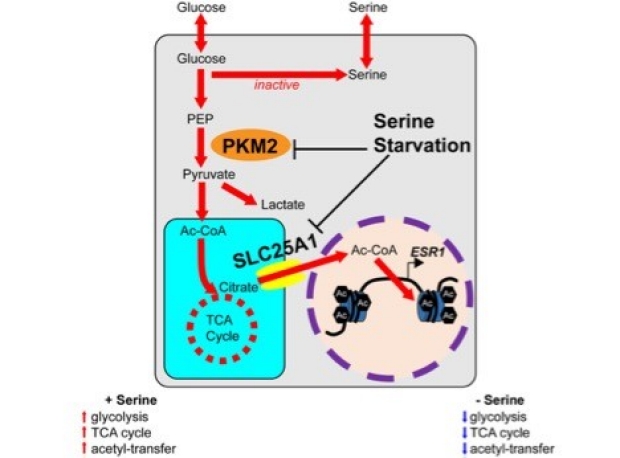
b. Serine Starvation Silences Estrogen Receptor Signaling through Histone Hypoacetylation
Loss of estrogen receptor (ER) pathway activity promotes breast cancer progression, yet how this occurs remains poorly understood. Here, we show that serine starvation, a metabolic stress often found in breast cancer, represses estrogen receptor alpha (ERα) signaling by reprogramming glucose metabolism and epigenetics. Using isotope tracing and time-resolved metabolomic analyses, we demonstrate that serine is required to maintain glucose flux through glycolysis and the TCA cycle to support acetyl-CoA generation for histone acetylation. Consequently, limiting serine depletes histone H3 lysine 27 acetylation (H3K27ac), particularly at the promoter region of ER pathway genes including the gene encoding ERα, ESR1. Mechanistically, serine starvation impairs acetyl-CoA-dependent gene expression by inhibiting the entry of glycolytic carbon into the TCA cycle and down-regulating the mitochondrial citrate exporter SLC25A1, a critical enzyme in the production of nucleocytosolic acetyl-CoA from glucose. Consistent with this model, total H3K27ac and ERα expression are suppressed by SLC25A1 inhibition and restored by acetate, an alternate source of acetyl-CoA, in serine-free conditions. We thus uncover an unexpected role for serine in sustaining ER signaling through the regulation of acetyl-CoA metabolism. (Li et al., PNAS. 2023)
Future directions:
1. Develop therapeutic strategies targeting mitochondrial serine and one-carbon unit metabolism.
2. Illuminate the molecular mechanisms by which SLC25A1 influences the compartmentalization of acetyl-CoA metabolism, impacting histone acetylation and transcriptional regulation.
3. Create epigenetic therapies to rejuvenate ER signaling in breast cancer.