To appear in: Oxford Handbook of Human Memory. M.J. Kahana & A.D. Wagner (Eds). Oxford University Press
9. Disorders of Memory
9.5 Basal Ganglia Diseases
Sephira G. Ryman, PhD, MS1,2, Assistant Professor
Kathleen L. Poston, MD, MS1, Associate Professor of Neurology and Neurological Sciences
1Neurology and Neurological Sciences, Stanford University School of Medicine, Stanford University, 300 Pasteur Dr. Room A343. MC-5235. Stanford, CA 94305 USA
2Translational Neuroscience, Mind Research Network, 1101 Yale Blvd., Albuquerque, NM 87106
Author emails:
Sephira Ryman: sryman@mrn.org
Kathleen Poston: klposton@stanford.edu
Abstract
The concept of multiple memory systems is based on the foundational studies that have dissociated the roles of the basal ganglia and medial temporal lobe structures in memory. Nondeclarative memory is heavily dependent on the basal ganglia, whereas declarative memory largely relies on the hippocampus and medial temporal lobe cortical structures. The nuances of these umbrella terms will be covered at length elsewhere in this book and we do not intend to suggest these systems are independent. However, this distinction is particularly useful when discussing memory functioning in basal ganglia disorders. For example, there is clear evidence for nondeclarative memory dysfunction in basal ganglia disorders, such as Parkinson’s disease. In addition, early in the disease course, individuals with Parkinson’s disease may exhibit poor declarative memory. The nature of these weaknesses may reflect poor retrieval processes secondary to fronto-striatal dysfunction or poor encoding suggestive of medial temporal lobe dysfunction. Notably, a declarative memory impairment may be an indicator of an emerging dementia process seen in both Parkinson’s disease dementia and Dementia with Lewy bodies, collectively referred to as Lewy body dementias. In this chapter, we first review the anatomy of basal ganglia disorders and focus initially on nondeclarative memory impairments in early Parkinson’s disease and other basal ganglia disorders. We then discuss the role of declarative memory dysfunction as a harbinger of a dementia syndrome and the complexity of interpreting memory impairments in light of the heterogeneity of neuropathological processes in Lewy body diseases.
Keywords
Basal ganglia, Parkinson’s disease, Huntington’s disease, Parkinson’s Disease Dementia, Dementia with Lewy bodies
Disorders of the Basal Ganglia
The basal ganglia are a cluster of subcortical nuclei that when damaged due to injury or neuropathological processes result in abnormal initiation and integration of movement. Basal ganglia components include the caudate nucleus, putamen, nucleus accumbens, globus pallidus, subthalamic nucleus, and the substantia nigra (Fahn, Jankovic, & Hallett, 2011). The caudate, putamen, and nucleus accumbens are collectively referred to as the striatum, which can be differentiated into the dorsal (caudate and putamen) and ventral striatum (nucleus accumbens; Figure 1). The substantia nigra is a midbrain dopaminergic nucleus which projects to the dorsal striatum. The dorsal striatum is implicated in movement, whereas the ventral striatum is more commonly associated with functions of the limbic system, such as emotions and reward processing. The globus pallidus is subdivided into the medial, or internal, component and the lateral, or external, component which are physically separated by descending white matter tracts from the cortex and are often abbreviated as GPi and GPe, respectively.
The basal ganglia plays a key role in the cortico-basal ganglia-thalamo-cortical loop, which involves functionally segregated connections between the cortex, basal ganglia, the thalamus and back to the cortex. The primary motor-related output from the basal ganglia arises from the GPi and the substantia nigra and projects to the ventrolateral and ventromedial nuclei of the thalamus via GABAergic fibers, which then project to the premotor cortex via glutamatergic fibers (Parent & Hazrati, 1995). Perturbations in this cortico-basal ganglia-thalamo-cortical loop, either due to neurodegenerative disorders like Parkinson’s disease or from lesions after stroke, most commonly result in either a failure to initiate movement or in excessive, involuntary movements, depending on where the insult occurs.
Box 1. Basal Ganglia Disorders
Hypokinetic Disorders
- Parkinson’s disease
- Dementia with Lewy bodies
- Progressive Supranuclear Palsy
- Corticobasal Syndrome
- Multiple System Atrophy
Hyperkinetic Disorders
- Huntington’s disease
- Essential Tremor
- Dystonia
- Restless Leg Syndrome
- Tics and Tourette syndrome
- Attention-deficit/hyperactivity disorder
Hypokinetic versus Hyperkinetic Disorders
There are two fundamental disorders of muscle activation in the absence of paralysis or weakness associated with these loops: hypokinetic and hyperkinetic movement disorders (Fahn, Jankovic, & Hallett, 2011; Box 1). Parkinson’s disorder is an example of a hypokinetic disorder characterized by slowness of movement (bradykinesia) and increased muscle tone (rigidity), which may be accompanied by a tremor at rest. Parkinson’s disease dementia and Dementia with Lewy bodies (both detailed at greater length below) are both dementia syndromes which exhibit dysfunction within the basal ganglia in addition to broader neuronal dysfunction. Hyperkinesias are characterized by excessive involuntary muscular activity that interfere with normal intended actions. Individuals with dystonia, for example, exhibit long muscle spasms with co-contraction of antagonist muscles which result in abnormal posturing. Huntington’s disease is an autosomal-dominant, hyperkinetic disorder which involves involuntary, irregular, purposeless, nonrhythmic, abrupt, rapid, unsustained movements that seem to flow from one body part to another (chorea) and dystonia. In addition to these movement disorders, the basal ganglia is also heavily involved in neuropsychiatric conditions such as attention deficit hyperactivity disorder, obsessive-compulsive disorder and Tourette syndrome (Fahn et al., 2011; Box 1).
Neuroanatomy of Parkinson’s Disease
The primary neuropathology that results in the cardinal symptoms of Parkinson’s disease (bradykinesia, rigidity, tremor) includes the misfolding and aggregation of α-synuclein in the SN and degeneration of nigrostriatal dopamine neurons. The SN is subdivided into the ventral pars reticulata (SNr) and the dorsal pars compacta (SNc); the latter is composed of dopaminergic neurons. Motor symptoms emerge after 50-70% loss of the dopaminergic neurons in the SNc (Damier, Hirsch, Agid, & Graybiel, 1999; Fearnley & Lees, 1991) resulting in dopaminergic denervation of the striatum. The SNc is further divided into the dorsal and ventral tier, with the loss of dopaminergic neurons occurring first in the caudal and ventrolateral tier (A9; Fearnley & Lees, 1991; Jellinger, 2012). The A9 dopaminergic neurons innervate primarily the motor and associative loops. However, α‐synuclein pathology and dopaminergic cell loss typically spreads to neighboring groups from A9 in Parkinson’s disease (A10; A8; Schulz-Schaeffer, 2010), impacting the function of the ventral tegmental area (VTA) in many cases (Alberico, Cassell, & Narayanan, 2015). These three cell groups project to the striatum in a topographic manner: A10 and A8 cell groups innervate the ventral striatum, inclusive of the nucleus accumbens, whereas the A9 cell group projects to the associative and motor parts of the striatum (caudate and putamen; Gaspar, Stepniewska, & Kaas, 1992; Williams & Goldman-Rakic, 1998).
In addition to the motor symptoms, Parkinson’s disease patients typically exhibit a variety of non-motor symptoms, an umbrella term that includes cognitive dysfunction, sensory abnormalities, behavioral changes, sleep disturbances, and autonomic dysfunction (Sauerbier, Jenner, Todorova, & Chaudhuri, 2016; Schapira, Chaudhuri, & Jenner, 2017). The presence and progression of both motor and non-motor symptoms is heterogeneous; however, both impact an individual’s ability to effectively engage in activities of daily living (Pfeiffer, 2016). Memory functioning in Parkinson’s disease is a particularly important area for future research as described in this chapter, there are various types of memory that are differentially affected in Parkinson’s disease.
Neuroanatomy of Huntington’s Disease
Huntington’s disease is characterized by extensive neuronal loss in the striatum and cerebral cortex. The mutation causing Huntington’s disease is an expansion of a CAG tract in exon 1 of the HTT gene (Fahn et al., 2011). The deleterious action of this mutation involves widespread areas of the brain, with certain areas more vulnerable to degeneration than others (U Rüb et al., 2016; Udo Rüb, Vonsattel, Heinsen, & Korf, 2015). Huntington’s disease is classically associated with caudate atrophy, which can be measured by the ratio of intercaudate to outer-table distances (Fahn et al., 2011). However, a reduction in the volume of the putamen measured by MRI may be a more sensitive index of neurologic dysfunction than caudate atrophy (Harris et al., 1992; Rosas et al., 2001), suggesting that the putamen is underdeveloped in Huntington’s disease or it is one of the earliest structures to atrophy. Due to the selective involvement of the basal ganglia early in the course of Parkinson’s disease and Huntington’s disease, they have been used to understand the cognitive neuroscience of nondeclarative memory processes.
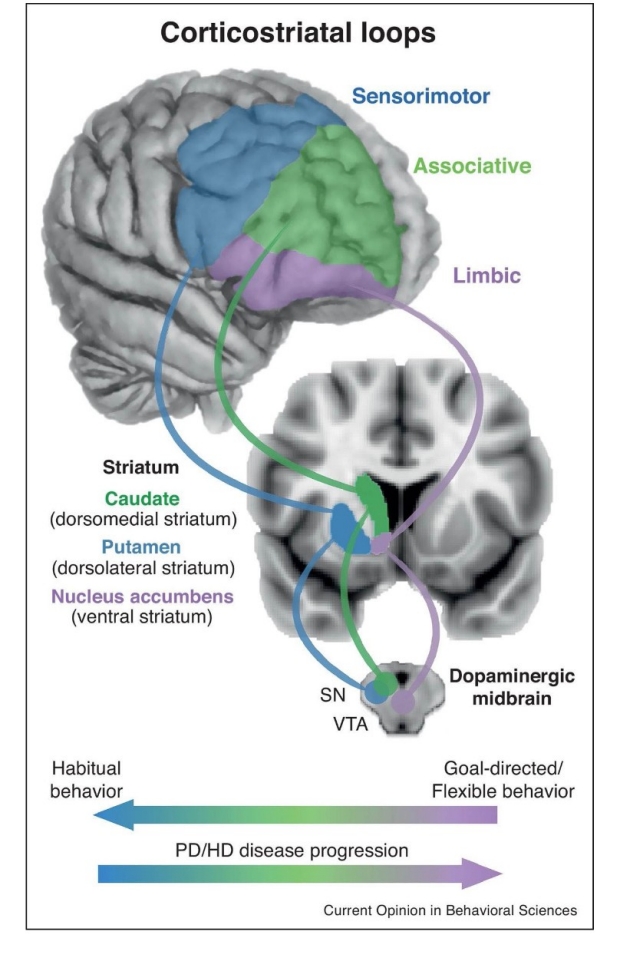
Figure 1. Simplified schematic of three fronto-striatal loops important for habitual and goal-directed behavior. (Bottom) Hypothesized progression of involvement of fronto-striatal loops as behavior develops from goal-directed to habitual and progression of striatal dysfunction. VTA, ventral tegmental area; SN, substantia nigra; PD, Parkinson’s disease; HD, Huntington’s Disease (Foerde, 2018).
Heterogeneous and Progressive nature of Parkinson’s disease and Huntington’s disease
There are several notable anatomical features of the basal ganglia and its connections that are important to understanding how disruption in this system can impact functioning. First, the cortico-basal ganglia-thalamo-cortical loop includes parallel-projecting, partially segregated reentrant loops that are subdivided into motor, associative (cognitive), and limbic (reward and emotions; Alexander, DeLong, & Strick, 1986; McHaffie, Stanford, Stein, Coizet, & Redgrave, 2005; Obeso, Rodriguez-Oroz, Stamelou, Bhatia, & Burn, 2014; Wiesendanger, Clarke, Kraftsik, & Tardif, 2004) collectively referred to throughout this chapter as the fronto-striatal loops (Figure 1). Depending on the disease process, there may be selective impairment in the motor loop (which is most commonly the case early in the course of Parkinson’s disease), however, it has become clear that both Parkinson’s disease and Huntington’s disease exhibit broader dysfunction, most notable as they progress. For instance, pathoanatomical studies of Huntington’s disease indicate that the neuroanatomical dysfunction is not confined to the caudate nucleus and putamen (U Rüb et al., 2016). Similarly, while Parkinson’s disease related changes often occur initially in the SNc, it is clear that the neuropathological degeneration (α‐synuclein) progresses to other regions of the brain (Heiko Braak, Ghebremedhin, Rüb, Bratzke, & Del Tredici, 2004). The progression of α‐synuclein beyond the SNc beyond the cortico-basal ganglia-thalamo-cortical loop corresponds to the development of symptoms associated with the function associated with the region impacted (e.g. progression to the limbic loop may lead to psychiatric dysfunction; H Braak, Rüb, Steur, Del Tredici, & De Vos, 2005). Importantly, the progression of the pathology to the cortical regions is associated with the development of a dementia syndrome and significant functional impairment.
Parkinson’s disease dementia and Dementia with Lewy bodies are distinct clinical syndromes that share the same underlying α‐synuclein neuropathology (Jellinger & Korczyn, 2018). The key distinction between these disorders is the timing of the onset of motor symptoms – when dementia develops after an established motor disorder that has been recognized for at least one year, it is diagnosed as Parkinson’s disease dementia, whereas when dementia develops prior to or at the same time as the motor disorder, it is considered Dementia with Lewy bodies (McKeith et al., 2017). For example, individuals with Parkinson’s disease can exhibit primarily motor symptoms for numerous years before developing a dementia process (Parkinson’s disease dementia). In contrast, Dementia with Lewy bodies typically exhibits a notable cognitive decline, highlighting a dementia process from the onset. Both Dementia with Lewy bodies and Parkinson’s disease dementia include cognitive impairment, parkinsonism (any condition that causes a combination of the movement abnormalities typically seen in Parkinson’s disease), visual hallucinations, and fluctuating attention. As the underlying neuropathology is similar in each disorder and the ‘one-year rule’ distinction is arbitrary (Postuma et al., 2016), we collectively refer to these disorders as Lewy body dementias in our subsequent discussions.
The nondeclarative memory processes discussed in the next section were primarily assessed early in the disease course of Parkinson’s disease and Huntington’s disease, and therefore likely are reflections of dysfunction in primarily the motor fronto-striatal loops (the putamen in Parkinson’s disease; putamen and caudate in Huntington’s disease) as opposed to ventral regions (nucleus accumbens; Aylward et al., 2004; Kish, Shannak, & Hornykiewicz, 1988; Péran et al., 2010; Péran, Nemmi, & Barbagallo, 2018). However, it is important to note that there is likely heterogeneity within these clinical samples, even early in the disease course. For instance, in Parkinson’s disease different subtypes of Parkinson’s disease that predict the rate of decline (Fereshtehnejad et al., 2015), highlighting the heterogeneous nature of the disorder, even early in the disease course. Often, studies of nondeclarative memory processes to not differentiate subtypes. We focus on Parkinson’s disease and Huntington’s disease in the non-declarative section with this caveat in mind. The latter section of the chapter reviews how dysfunction in Parkinson’s disease and Lewy body dementias impacts declarative memory function.
Nondeclarative memory processes in basal ganglia disorders
There is an enormous amount of prior research that investigates nondeclarative memory dysfunction in basal ganglia disorders, particularly with respect to Parkinson’s disease and Huntington’s disease. As there are several parallel lines of research that characterize nondeclarative memory processes in a variety of ways, an exhaustive review is not possible within this chapter. One of the most parsimonious accounts of nondeclarative memory dysfunction in Parkinson’s disease indicates that patients are impaired on tasks that involve gradual learning of stimulus-response associations, often referred to as habit learning (Foerde & Shohamy, 2011; Packard & Knowlton, 2002). More recently, it is appreciated that the basal ganglia is not limited to habit learning, but also goal-directed motivated behaviors that direct aspects of habit learning and related processes (Foerde, 2018).
Here we aim to briefly discuss procedural motor learning and reinforcement learning as a means to illustrate how these abilities are disrupted in Parkinson’s disease and Huntington’s disease. Procedural learning is a form of gradual learning of stimulus-response associations, which refers to the ability to acquire motor or cognitive skills gradually through practice (Cohen & Squire, 1980; see also, Chapter 2.4). Implicit motor skill learning is commonly examined using the serial reaction time task (Nissen & Bullemer, 1987). The serial reaction time task is a choice reaction time task that requires participants to respond quickly to a visual stimulus presented at one of several different spatial locations. The task manipulates ‘sequence-specific learning’, that is the improvement of responding due to the repetition of the sequence of stimuli. ‘Non-specific learning’ is a general process whereby increased response speed (or accuracy) is observed over the course of performances.
Serial reaction time task performance is associated with striatal engagement in combination with the premotor cortex and supplementary motor area (Grafton, Hazeltine, & Ivry, 1995; Rauch et al., 1997). Individuals with Parkinson’s disease or Huntington’s disease (Knopman & Nissen, 1991) exhibit impairments in implicit sequence learning, though these findings are not consistent across studies (Brown, Redondo-Verge, Chacon, Lucas, & Channon, 2001; Jackson, Jackson, Harrison, Henderson, & Kennard, 1995; Stefanova, Kostic, Ziropadja, Markovic, & Ocic, 2000). Reviews of the literature suggest that Parkinson’s disease patients are less efficient, but able to acquire procedural knowledge of a learning sequence. The impairment in this ability is more pronounced in individuals with more severe clinical syndromes, and is not due to dopaminergic medication. Impaired procedural motor learning does not affect functional status (Muslimović, Post, Speelman, & Schmand, 2007; Gobel et al., 2013; Nieuwboer, Rochester, Müncks, & Swinnen, 2009).
The serial reaction time task does not include feedback on the participant’s responses, but rather responses are prompted by externally signaled stimuli. Similar to the serial reaction time task, a related task, the probabilistic classification task has been used to examine gradual learning of stimulus-response associations, with the addition of providing response-contingent feedback to participants. The probabilistic classification task requires that participants use trial-and-error feedback to learn to predict categorical outcomes (most commonly weather predictions: sun or rain) based on four different visual cues. The relationship between cues and outcomes is probabilistic (i.e. the cue predicts an outcome only some of the time) to make improved performance dependent on gradual implicit learning (Knowlton, Mangels, & Squire, 1996; Knowlton & Squire, 1993). Early studies highlighted that individuals with amnestic memory deficits (resulting from damage to the medial temporal lobe) were able to complete the probabilistic classification task with no issues, whereas Parkinson’s disease and Huntington’s disease patients exhibited poor classification learning on the probabilistic classification task (Knowlton et al., 1996; Knowlton, Squire, & Gluck, 1994; Shohamy et al., 2004).
We highlight that these nondeclarative memory processes are rarely entirely implicit, but often driven by goal-directed or habitual control processes, which is reflected in the heavily integrated fronto-striatal systems (Redgrave et al., 2010). More recently, the focus has shifted from understanding habitual versus goal-directed control of behavior, and there has been some inconsistent reports with respect to Parkinson’s disease. In two studies, habit learning was intact and, instead, goal-directed behavior was impaired (de Wit, Barker, Dickinson, & Cools, 2011; Sharp, Foerde, Daw, & Shohamy, 2015). This has raised concerns that earlier studies using the probabilistic classification task capture a deficit in goal-directed learning rather than habit learning (Foerde, 2018) and highlights the necessity of precise operationalization of cognitive neuroscience tasks and general issues with regard to the ontology of learning and memory processes (Poldrack & Yarkoni, 2016). Furthermore, it is clear that basal ganglia disorders also exhibit broader executive functioning deficits such as poor internal control of attention, working memory, set shifting, planning, inhibitory control, dual task performance, and a range of decision‐making and social cognition functions (Dirnberger & Jahanshahi, 2013).
It is not clear how independent the gradual learning of stimulus-response associations in the probabilistic classification task and serial reaction time task –– classically characterized as nondeclarative memory tasks –– are from prototypical executive functioning tasks typically utilized in clinical settings, such as the Wisconsin Card Sorting Task, that demonstrate medium to large effect sizes in Parkinson’s disease (Berg, 1948; Heaton, Chelune, Talley, Kay, & Curtiss, 1993; Lange, Brückner, Knebel, Seer, & Kopp, 2018). The Wisconsin Card Sorting Task is similar to the probabilistic classification task, as it requires participants to sort cards based on dimensional categories (color, shape, or number) that are not revealed to the individual. Based on the examiner’s feedback, the individual is expected to test rules and identify the correct category. Unlike the probabilistic classification task, it is assumed that the sorting categories are explicitly determined by the individual. Once they have identified the category and completed a predefined number of consecutive correct placements, the task rule changes, resulting in negative feedback. Participants are given a set number of cards to sort and at the end of the task, the total number of correctly identified sorting categories is one of the primary measures examined. Parkinson’s disease patients typically correctly complete a fewer number of categories relative to healthy controls. Additionally, Parkinson’s disease patients exhibit particular difficulty adapting to the change in rule, resulting in what are referred to as perseverative errors (e.g. continuing to pursue a previously correct category that is now incorrect). These deficits were medium to large in effect size and are more pronounced in Parkinson’s disease patients that are tested off dopaminergic medications medications. The extensive research on the Wisconsin Card Sorting Task provides further support that the non-declarative memory functions in Parkinson’s disease are possibly related to impaired goal-directed behavior.
In the context of the assumed early neurodegeneration in Parkinson’s disease (initially affecting SNc but possibly progressing to the VTA, depending on the individual case), it is important to map these cognitive changes more directly onto the underlying circuitry. For example, the ability to learn stimulus-reward associations on the basis of reward prediction errors depends on the mesolimbic dopaminergic system (Schott et al., 2007), which may be a reflection of SNc or VTA degeneration. An understanding of these relationships will provide further insight into the habitual versus goal-directed changes observed in Parkinson’s disease (Redgrave et al., 2010). It is also important to account for the progressive nature of diseases like Parkinson’s disease. While early in the disease course, there may be circumscribed deficits in the nigrostriatal system, however, this is likely not consistent across individuals and across time as the underlying neuropathology progresses. Incorporation of imaging and other biomarkers will be useful to better understand these systems.
Summary of Nondeclarative Memory Processes in basal ganglia disorders
These studies primarily utilize Parkinson’s disease and basal ganglia disorders as a disease model to further understand the neural mechanisms of memory. A review of the nondeclarative memory processes here highlights that a wealth of cognitive processes and behavioral phenomena have been identified, and there is considerable evidence of dysfunction in stimulus-response learning. However, there is also variability in how these processes are disrupted in basal ganglia disorders, which may be a reflection of progression of underlying disease processes. This is also likely related to the continually evolving ontology of nondeclarative memory processes which has shed light on various aspects of dysfunction, but fail to present a general model of basal ganglia dysfunction in Parkinson’s disease and Huntington’s disease (Eisenberg et al., 2018).
One interesting aside is that nondeclarative memory functioning is not typically examined clinically, but may be a useful way to assess aspects of dysfunction. There is certainly several opportunities for future research to understand the differential involvement of the dopaminergic innervation from the SN and VTA in nondeclarative memory processes. Dysfunction in aspects of learning processes are likely closely linked with both the early cognitive deficits in executive functioning as well as the observed changes in psychiatric symptoms in Parkinson’s disease, such as lack of motivation, anxiety, and depression.
Declarative Memory in basal ganglia disorders
Declarative or explicit memory involves the acquisition of facts and events with conscious awareness of the learned information. Declarative memory is commonly assessed using verbal and visuospatial tests that require the individual to recall and recognize previously learned material (Eichenbaum, 2000; Tulving & Markowitsch, 1998). Declarative memory is associated with the hippocampus and medial temporal lobe cortical structures (Squire & Zola-Morgan, 1991). Early dissociations highlight the relatively distinct nature of the declarative memory system relative to the nondeclarative system, with Alzheimer’s disease patients exhibiting an amnestic syndrome compatible with dysfunction of the hippocampus and temporal cortex (see Chapter 9.2). Parkinson’s disease and Huntington’s disease patients, in contrast, typically exhibit inefficient planning of memory processes related to the fronto-striatal dysfunction early in the disease course (Kramer, Levin, Brandt, & Delis, 1989; Pillon, Deweer, Agid, & Dubois, 1993).
Notably, there is variability in the memory profile observed in Parkinson’s disease patients that is likely a reflection of the heterogeneity of the disorder and understanding this heterogeneity will be critical to accurately characterizing disease processes in Parkinson’s disease and Lewy body dementias.
Verbal list learning tests, such as the California Verbal Learning Test (D. Delis, Kramer, Kaplan, & Ober, 1987) and the Hopkins Verbal Learning Test (Brandt, 1991) have been particularly useful to differentiate the memory profiles of individuals with Parkinson’s disease, Huntington’s disease, and Alzheimer’s disease (Helkala, Laulumaa, Soininen, & Riekkinen, 1988; Jacobs et al., 1995; Kramer et al., 1989). Patients are presented with a list of words from several categories (e.g. vegetables, animals) across several trials (3 for the Hopkins Verbal Learning Test; 5 for the California Verbal Learning Test). Patients are asked to recall this information following a delay (most commonly 20 minutes), with the number of words recalled referred to as free recall. Additionally, patients are asked to determine if individually presented words were on the word list by responding “yes” or “no”. Recognition discrimination is typically quantified as the difference between true hits (correct “yes”) and false positives (incorrect “yes”). As free recall requires both effective encoding of the information (mediated by hippocampal/medial temporal cortical function) and retrieval processes (mediated by fronto-striatal function), whereas recognition discrimination is less reliant on fronto-striatal processes, these combination of these measures can provide information regarding the fidelity of these two systems.
For example, the classic memory profile associated with subcortical neurodegenerative conditions, such as Parkinson’s disease and Huntington’s disease (Butters, Wolfe, Martone, Granholm, & Cermak, 1985), consists of poor delayed free recall with relative sparing of recognition performance, referred to as a retrieval deficit consistent with fronto-striatal dysfunction. In contrast, Alzheimer’s disease patients exhibit poor delayed free recall and impaired recognition, indicating an encoding deficit (D. C. Delis et al., 1991; Heindel, Salmon, Shults, Walicke, & Butters, 1989; Helkala et al., 1988; Jacobs et al., 1995; Kramer et al., 1989). The encoding deficit is often referred to as an amnestic deficit and is typically associated with hippocampal dysfunction (Dubois & Albert, 2004). While this pattern holds in Parkinson’s disease, generally, there are reports of normal performance on both free recall and recognition as well as a quarter or less exhibiting an encoding deficit (Filoteo et al., 1997; Weintraub, Moberg, Culbertson, Duda, & Stern, 2004). A recent review indicates that there is a paucity of longitudinal neuropsychological data available to even begin to understand the nature and progression of cognitive deficits in Parkinson’s disease (Roheger, Kalbe, & Liepelt-Scarfone, 2018).
While this chapter focuses on memory processes, we briefly discuss the diagnosis of mild cognitive impairment in Parkinson’s disease as it has only recently been defined and a comprehensive understanding of the etiology of memory deficits in Parkinson’s disease will likely be critical to appropriately characterize mild cognitive impairment and its prognostic utility. In Alzheimer’s disease, the diagnosis of mild cognitive impairment was developed to capture prodromal patients, or patients in a clinical condition between normal aging and Alzheimer’s disease in which individual experience memory loss to a greater extent than one would expect for age (Petersen et al., 2001). The neuropsychological profile early in the course of Alzheimer’s disease is relatively circumscribed and maps onto the progression of the underlying tau neuropathology (initial encoding or amnestic memory impairments correspond to tau tangles within the medial temporal lobes, which then progresses to affect the semantic language networks in the broader temporal lobe (Bejanin et al., 2017; Bondi et al., 2008)). A clear amnestic memory deficit is present early in the disorder, years before the development of the dementia syndrome (Dubois & Albert, 2004). Developing a similar diagnostic entity in Parkinson’s disease has proven to be more challenging, as it is not clear to what extent memory dysfunction relative to other cognitive domain functions are indicative of the progression of underlying pathology.
The diagnosis of mild cognitive impairment was recently formally defined in Parkinson’s disease (Litvan et al., 2012). There are two levels of assessment based on the availability or practicality of a comprehensive neuropsychological assessment. Level 1 is based on the use of a global cognitive abilities scale [e.g. Montreal Cognitive Assessment (MoCA); (Dalrymple-Alford et al., 2010)], resulting in possible Parkinson’s disease with mild cognitive impairment, whereas Level II assessment is based on a more comprehensive assessment on at least two tests for each of the five commonly assessed cognitive domains (attention and working memory, executive function, language, memory, and visuospatial function), and impairment should be present on at least two tests, either within a single cognitive domain or across different cognitive domains. Importantly, intact activities of daily living are required for a diagnosis of Parkinson’s disease with mild cognitive impairment, as the development of impairments in activities of daily living is an indicator of a dementia process.
Approximately 20-25% of Parkinson’s disease patients meet criteria for mild cognitive impairment at diagnosis, over 40% of patients develop mild cognitive impairment within 6 years (Pigott et al., 2015), and 80% of those who survive two decades progress to Parkinson’s disease dementia (Aarsland & Kurz, 2010; O'Callaghan & Lewis, 2017). In one study, of the 25.8% of Parkinson’s disease patients that exhibited mild cognitive impairment across 8 separate cohorts, memory impairment was most common, followed by impairment in visuospatial and attention/executive abilities (Aarsland et al., 2010). In a separate study of 128 Parkinson’s disease patients with mild cognitive impairment subtypes, attentional/executive and visuospatial abilities were the most frequently impaired domains, but approximately 42% of the patients with mild cognitive impairment exhibited either a memory-only or memory-plus impairment in another domain (Goldman, Weis, Stebbins, Bernard, & Goetz, 2012). It is important to note that the term “amnestic memory impairment” has been used inconsistently in the Parkinson’s disease literature. As noted, an amnestic or encoding memory impairment is associated with hippocampal dysfunction and refers to a specific pattern of memory performance. In the Parkinson’s disease literature, often any memory impairment is often referred to as an amnestic deficit, which limits the ability to differentiate how different memory profiles may predict emerging dementia processes.
While the formal definition of MCI has been useful in identifying individuals who have begun to experience cognitive difficulties in Parkinson’s disease, it is important to highlight that Parkinson’s disease with mild cognitive impairment does not reflect a distinct clinical entity, but likely reflects a mixture of pathophysiological mechanisms (Wen, Chan, Tan, & Tan, 2017). For example, it is thought that α‐synuclein aggregation typically begins in the caudal brainstem and progresses rostrally (Heiko Braak et al., 2004; Dickson, 2018); the progression is found to correspond to increased cognitive impairment (H Braak et al., 2005), but the nature of these differences depends on the stage of progression and whether co-morbid pathology is present. The stages of pathology have been formally characterized, referred to as the Braak Lewy body stages (Heiko Braak et al., 2004), with more recent updates including brainstem and limbic predominant stages (Beach et al., 2009). Parkinson’s disease dementia patients exhibit higher cortical and limbic α‐synuclein pathology than Parkinson’s disease patients (Hurtig et al., 2000; Irwin et al., 2012; Kempster, O’Sullivan, Holton, Revesz, & Lees, 2010). The burden of cortical α‐synuclein is linked to a decline in cognitive performance as well (Aarsland, Andersen, Larsen, & Lolk, 2003; H Braak et al., 2005; Mattila, Rinne, Helenius, Dickson, & Röyttä, 2000). To summarize, as Lewy body pathology progresses to limbic and cortical regions, dementia develops and both declarative and nondeclarative memory processes are disrupted.
This is further complicated by the fact that cognitive impairment and dementia in these disorders develop not only by α-synuclein-related neurodegeneration but also by the presence of heterogeneous neuropathology. For example, Parkinson’s disease patients often exhibit Lewy body as well as Alzheimer disease neuropathologic change at death (Irwin et al., 2012). For a detailed review of Alzheimer’s pathology, please see chapter 9.2. Briefly, there are two important neuropathological processes in Alzheimer’s disease, 1) senile plaques, which are extracellular deposits of amyloid-β (Aβ) peptides and 2) neurofibrillary degeneration, which is best exemplified by neurofibrillary tangles (Montine et al., 2012). Similar to α‐synuclein, a staging of neuropathology has been formally described in Alzheimer’s disease (Heiko Braak, Alafuzoff, Arzberger, Kretzschmar, & Del Tredici, 2006). It is not known whether the Alzheimer disease neuropathologic change follows a similar pattern when present in the context of α‐synuclein pathology. While there are differences in the cohorts examined, one study highlighted the following patterns of neuropathology reported in Parkinson’s disease dementia: 38% exhibit predominant synucleinopathy (Braak Lewy body Stages 5-6), 59% exhibit predominant synucleinopathy with Aβ deposition (Braak amyloid Stages B-C) with minimal or no cortical tau deposition, and 3% exhibit synucleinopathy and Aβ deposition with at least moderate neocortical tauopathy (Braak tau Stages 5-6; Kotzbauer et al., 2012).
While the progression of α-synuclein pathology to the medial temporal lobes likely contributes to memory impairment in Lewy body dementias (Adamowicz et al., 2017), it is unclear to what extent Alzheimer’s disease neuropathological change plays a role. Several indicators of Alzheimer’s disease neuropathological change have been found to predict cognitive decline in Parkinson’s disease, such as CSF tau and tau/Aβ42 (Liu et al., 2015), CSF Aβ42 (Leaver & Poston, 2015), and positron emission tomographic (PET) amyloid measured with Pittsburgh compound B retention (Gomperts et al., 2013). The presence of tau and Aβ based on PET imaging did not explain mild cognitive impairment in a small study of Parkinson’s disease (Winder et al., 2018). However, this may be related to heterogeneity in cognitive dysfunction –– attention/working memory, episodic memory, and executive functioning were the primary domains affected, but to a different extent across individuals. To fully understand these processes, diagnostic and progression biomarkers of α-synuclein and Alzheimer’s pathology will be crucial to differentiate the impact of these underlying neuropathological processes on declarative memory performance in Parkinson’s disease and Lewy body dementias. Understanding these relationships will have relevance for predicting progression in these disorders as well as aid in the development of disease-modifying interventions.
Summary
There is considerable evidence that “memory” involves a set of distinct systems that are functionally and neurobiologically dissociable based on foundational studies inclusive of basal ganglia disorders (Cohen, Poldrack, & Eichenbaum, 1997; Gabrieli, 1998; Poldrack & Foerde, 2008; Squire & Zola, 1996). One of the most parsimonious accounts of nondeclarative memory dysfunction in Parkinson’s disease indicates that patients are impaired on tasks that involve gradual learning of stimulus-response associations, often referred to as habit learning. More recent investigation highlights the importance of goal-directed learning in Parkinson’s disease and indicates the need for further understanding of the dynamic processes of the fronto-striatal system not only in stimulus-response learning, but rather in tasks that also require goal- directed feedback.
The latter half of this chapter discussed the utility of differentiating between encoding and retrieval deficits to further understand the nature of declarative memory deficits in Parkinson’s disease and Lewy body dementias. The presence of a retrieval deficit is thought to reflect dysfunction in the fronto-striatal system, and can occur in the context of preserved medial temporal cortical function in Parkinson’s disease and Lewy body dementias. In contrast, an encoding deficit is associated with hippocampal and medical temporal cortical dysfunction. Both α-synuclein and Alzheimer’s disease neuropathological change may contribute to the encoding impairment that is inconsistently observed in Parkinson’s disease and Lewy body dementias. The advent of biomarkers will undoubtedly facilitate a better understanding of the relationship between underlying neuropathological processes and disrupted declarative memory in Parkinson’s disease and Lewy body dementias.