A common signaling pathway unites diverse fibrotic diseases in humans, Stanford researchers have found. An antibody called anti-CD47, which is being tested as an anti-cancer agent, reverses fibrosis in mice.
April 17, 2017 - By Krista Conger
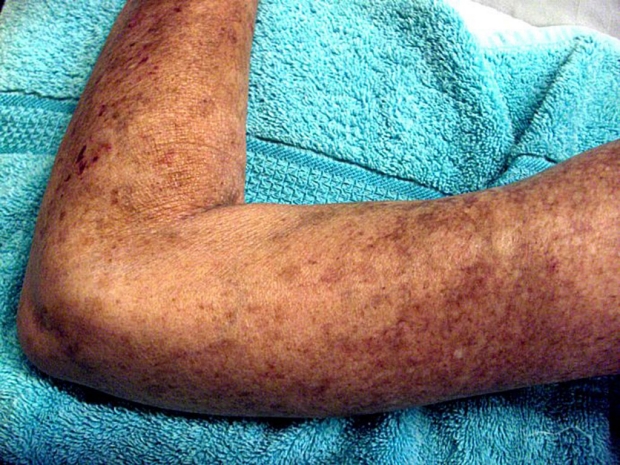
Scleroderma is an autoimmune disease that causes scarlike thickening of the skin and internal organs. Stanford researchers believe they've found a pathway that links a variety of diseases like scleroderma, fibrosis and liver cirrhosis.
Wikimedia Commons
Researchers at the Stanford University School of Medicine have identified a pathway that, when mutated, drives fibrosis in many organs of the body.
The pathway underlies what have been considered somewhat disparate conditions, including scleroderma, idiopathic pulmonary fibrosis, liver cirrhosis, kidney fibrosis and more, the researchers found. These diseases are often incurable and life-threatening.
Importantly, the researchers were able to reverse lung fibrosis in mice by administering an antibody called anti-CD47 now being tested as an anti-cancer treatment.
“The variety of diseases caused by overproduction of fibroblasts has made finding a common root cause very challenging, in part because there has been no good animal model of these conditions,” said Irving Weissman, MD, professor of pathology and of developmental biology. “Now we’ve shown that activating a single signaling pathway in mice causes fibrosis in nearly all tissues. Blocking the CD-47 signal, which protects cancer cells from the immune system, can also ameliorate these fibrotic diseases even in the most extreme cases.”
The researchers hope their findings will lead to the development of a reliable treatment of many types of fibrotic diseases. They are also planning to investigate whether the anti-CD47 antibody could be an effective treatment for people with fibrosis.
A study describing the research was published online April 17 in the Proceedings of the National Academy of Sciences. Weissman, who directs Stanford’s Institute for Stem Cell Biology and Regenerative Medicine and the Ludwig Center for Cancer Stem Cell Research and Medicine, is the senior author. Gerlinde Wernig, MD, assistant professor of pathology, is the lead author.
When injury response goes astray
Fibrosis occurs when the body’s normal response to injury goes astray. An overenthusiastic or inappropriately timed proliferation of cells called fibroblasts, which make up the connective tissue surrounding and supporting all of our organs, can lead to many devastating diseases. Until now, it’s not been clear whether these diseases share a common biological pathway.

Gerlinde Wernig
The researchers were building upon previous work by Wernig on a condition called myelofibrosis, or fibrosis of the bone marrow. In a mouse model she developed, she had found that fibroblasts were producing unusually high levels of an important signaling molecule called c-Jun. C-Jun is a transcription factor that drives the production of many proteins involved in critical cellular processes. It’s been implicated in many types of human cancer.
In the current study, Wernig investigated c-Jun expression levels in 454 biopsied tissue samples from patients with a variety of fibrotic diseases. She found that in every case the fibroblasts from the patients with fibrosis expressed higher levels of c-Jun than did control fibroblasts collected from people with nonfibrotic conditions.
“We found that c-Jun is not just over-expressed, but it’s also highly activated,” Wernig said. “We wondered if its activity is necessary to maintain the disease.”
Blocking the expression of c-Jun in laboratory-grown lung fibroblasts collected from people with idiopathic pulmonary fibrosis substantially decreased the proliferation of these cells, but not of lung fibroblasts collected from people without fibrosis, Wernig said. Furthermore,
mice genetically engineered to overexpress c-Jun in all their body’s tissues developed fibrosis in nearly every organ, including lung, liver, skin and bone marrow. Finally, she also found an intriguing link to past work from the Weissman lab.
‘A unifying mechanism’
“We found that c-Jun overexpression and over-activation is a unifying mechanism in many types of fibrosis,” Wernig said. “But an even more exciting part of the story is the fact that we observed that the diseased, c-Jun-expressing fibroblasts are surrounded by immune cells called macrophages. This is reminiscent of what’s often seen in human cancers.”

Irving Weissman
Over the past eight years, researchers in Weissman’s laboratory have shown that many human cancers evade the immune system by expressing high levels of a protein called CD47 on their surfaces. Blocking this protein with an anti-CD47 antibody restores the ability of the macrophages to gobble the cancer and has proven to be a promising treatment in animal models of the disease. Anti-CD47 antibody is currently undergoing a phase-1 clinical trial in humans with advanced solid tumors.
“Like in cancer, these fibroblasts are proliferating excessively beyond what should be their natural limit,” Weissman said. “We therefore wondered whether they are also expressing the ‘don’t eat me’ signal on their surfaces to protect them from the immune system.”
When Wernig treated mice with c-Jun-induced lung fibrosis with daily injections of anti-CD47 antibody, the animals exhibited significantly better lung function, lived longer than their peers and cleared the fibrosis.
The researchers plan to investigate whether any patients in the phase-1 trial of the anti-CD47 antibody also suffered from any fibrotic conditions. If so, they are eager to learn whether they experienced any relief as a result of participating in the trial.
“We have hit upon something unique in this study,” Wernig said. “We identified a highly activated pathway that causes fibrosis in many tissues in mice, and we’ve showed that treating the animals with an anti-CD47 antibody reverses the fibrosis. We’re hopeful that this could be a potential treatment for people with many types of fibrotic conditions.”
This study shows once again how basic science investigations in one field can lead to advances in what appeared to be unrelated diseases.
Wernig also tested inhibitors of other genes activated by c-Jun in the abnormal fibroblastic cells, and inhibitors of two pathways also reduced the fibrotic lesions.
“This study shows once again how basic science investigations in one field can lead to advances in what appeared to be unrelated diseases,” Weissman said. “Here, our studies of human cancer have led to the discovery of the mechanisms of how other ‘dangerous’ cells in fibrosis escape removal by the body’s scavenger cells. It shows how important it is to develop appropriate animal models of human diseases and then to use those models to identify disease-specific pathways that can be targeted.”
Weissman is the Virginia & D.K. Ludwig Professor for Clinical Investigation in Cancer Research. He is a member of Stanford’s Bio-X, Cardiovascular Institute and Cancer Institute.
Other Stanford co-authors are postdoctoral scholars Shih-Yu Chen, MD, PhD, and Lu Cui, PhD; former undergraduate student Camille Van Neste; graduate student Jonathan Tsai; professor of pathology Neeraja Kambham, MD; professor of pathology and of pediatrics Hannes Vogel, MD; professor of pathology Yaso Natkunam, MD, PhD; and professor of microbiology and immunology Garry Nolan, PhD.
The research was supported by the National Institutes of Health (grants U01HL099999, R01CA86017, U19AI057229, U19AI100627, R33CA183654, R33CA0183692, R01GM10983601, R01CA184968, R01CA19665701, R21CA183660, R01NS08953301, 5UH2AR067676 and R01HL120724); the Department of Defense; the Food and Drug Administration; the Gates Foundation; the Virginia and D.K. Ludwig Fund for Cancer Research; the Stanford Cancer Institute; the Stanford Physician Scholar Society; the Institute for Immunity, Transplantation and Infection; Northrop-Grumman Corp.; Novartis; Pfizer; and Juno Therapeutics.
Weissman is the founder of Forty Seven Inc., which is exploring ways to use immunotherapy like the anti-CD47 antibody to fight cancer. A patent titled “Antifibrotic activity of anti-CD47 blockade” has been filed by the researchers.
Stanford’s Department of Pathology also supported the work.
-
 Krista CongerKrista Conger is a senior science writer in the Office of Communications. Email her at kristac@stanford.edu.
Krista CongerKrista Conger is a senior science writer in the Office of Communications. Email her at kristac@stanford.edu.
About Stanford Medicine
Stanford Medicine is an integrated academic health system comprising the Stanford School of Medicine and adult and pediatric health care delivery systems. Together, they harness the full potential of biomedicine through collaborative research, education and clinical care for patients. For more information, please visit med.stanford.edu.

