Frequently Asked Questions
What kind of tubes should I use/what information should be included on the label?
We recommend 1.5ml low-binding DNA micro centrifuge tubes. Do not use 0.5ml, 2ml, or screw cap micro centrifuge tubes.
We prefer printed labels or legible handwriting that includes the following information on the side of the tube:
- DNA concentration (ng/ul)
- Submitter name or initials
- Unique library ID, meaning you have not used this ID before in previous submissions
Please write or print the unique library ID on the lid of the sample.

How should I package my libraries?
Use a small plastic bag to contain your tubes and a printed copy of your submission form.
If you have two libraries with different Illumina platforms, please send us two independent submission forms, one for each request.
How do I fill out the Excel spreadsheet?
Do not erase, format or change the fields on the Excel form.
You must include all the information required for each library.
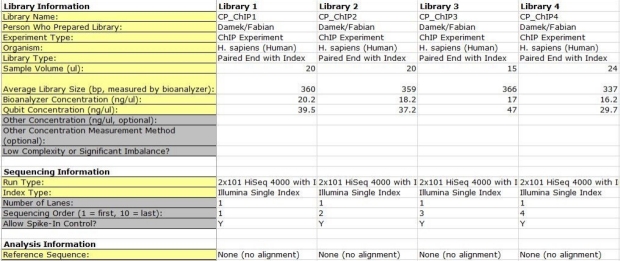
Please double check your index sequences to make sure they are correct and are the same length.
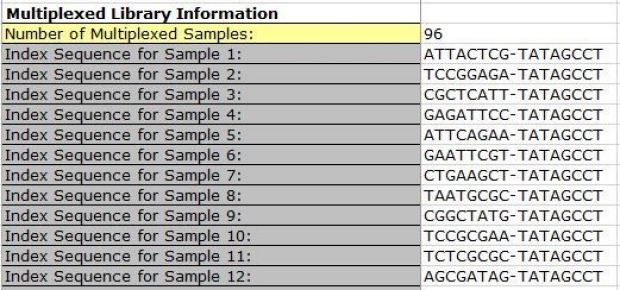
The indexes must be the same number of nucleotides in i7 and i5. If your run is a dual index, please include the i7 and i5 nucleotide sequences separated by a dash “-“, for each library.

Do I need to include other types of documentation with my submission?
Yes, it is a requirement to include the BioAnalyzer information for the pools. Do not send Bioanalyzer information for each individual library in the pool. We prefer to have the Bioanalyzer of the pool. Please email it with your submission form.
What does low nucleotide diversity or low complexity in a library mean?
Nucleotide diversity is the equal proportions of A, C, G and T nucleotides at each base composition in a sequencing library.
Illumina platforms require sufficient nucleotide diversity for effective template generation.
Sequencing a low diversity library is challenging, but can be accomplished by introducing a proportional amount of spike-in PhiX control in the library prior to the denaturation step. The lower the nucleotide diversity, the more spike-in PhiX control will be introduced into the library. The purpose is to balance the diversity of your library using the PhiX control. Another alternative is to use a well balanced library to spike-into the low diversity nucleotide libraries.
At the end, your library will be sequenced with optimal QC30 scores and higher percentage of passing filter reads. You will also see a percentage of PhiX control reads in the sequencing results that easily can be filtered out during data analysis.
How do I know if my library has low nucleotide diversity or low complexity?
The following categories often have low nucleotide diversity and/or low complexity:
- Libraries which are generated without random fragmentation
- Libraries produced through amplicon generation
- Bisulfite libraries
- Single amplicon generation protocols
- Other custom construction that may have flanking construction with same base composition
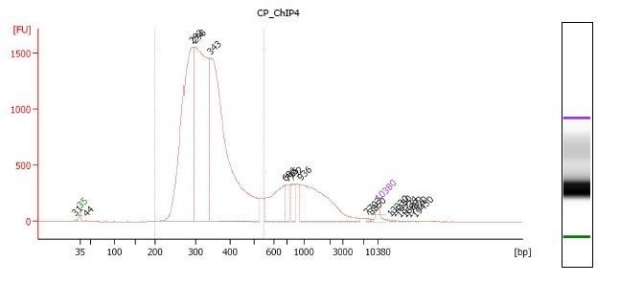
How do I determine the fragment size of my library?
We prefer you use a Bioanalyzer or a similar instrument to estimate the fragment size of your library. Please see the figure to help you to estimate your library fragment size. If you use a Bioanalyzer, use the region table on the area where the majority of your fragments are gathered.


How does the DGAC perform quality control of my library?
The DGAC uses a qPCR assay (KAPA) to functionally quantify your libraries. The final DNA concentration is determined based on this parameter as well as the evaluation of your BioAnalyzer information.
The Bioanalyzer information helps us to verify that your library fragment size is calculated correctly. It can also give us an idea of how much adapter dimer contamination is in your library.
Can you accept libraries with adapter dimer contamination?
The DGAC will accept libraries with less than 0.5% of adapter dimer contamination. If you are over this threshold, please do an AMPure bead clean up to remove the adapter dimers.
It is very important to minimize the percentage of primer dimer molecules in your libraries to less than 0.5% because adapter dimers are very competitive molecules, especially if you want to sequence your libraries in an Illumina HiSeq4000 platform.
How can I clean up my libraries if they have more than 0.5% adapter dimers?
You can use an AMPure bead clean up protocol or gel purification.
A protocol for using AMPure XP beads can be found here (PDF)
How does the percentage of adapter dimers affect my sequencing results?
As you increase the adapter dimer contamination in your final libraries, you increase the chance that adapter dimers will compete with your library fragments. With just a 5% adapter dimer contamination you will end up with 50% of your reads being an adapter dimer sequence. It is especially important to minimize the adapter dimer contamination for Illumina Hiseq4000 because of the Exclusion Amplification chemistry during cluster generation.
How will I receive my data?
All the data generated from the DGAC is accessible through DNAnexus, even if you requested no alignment and only fastq files.
If you are a new DGAC customer make sure to create an account, which is free only for the first month. Please make sure to download the files you need because you will only have access to your data for 30 days, starting from when you received an email notification.
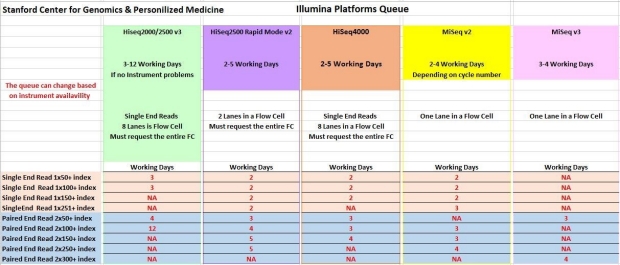
How long is the sequencing queue?
We try to sequence your libraries as soon as possible. The sequencing queue time depends on which Illumina platform you use. Please check the table below for an idea of the sequencing queue times for each Illumina platform.
How many reads should I expect for different Illumina platforms?
It is very important to plan your experiment ahead to make sure you cover an acceptable amount of reads. Please see the table below with all of the types of runs across different Illumina platforms. The data in this table is an estimation depending on the quality and performance of your library. At DGAC we make an effort to give you the maximum amount of reads with the highest quality possible.

How can I calculate how much coverage my experiment needs?
Illumina has a calculator that will help you to plan your experiment that chooses the right Illumina platform for your needs.