Stopping Pulmonary Arterial Hypertension Progression
by Amanda Chase, PhD
March 9, 2023
Pulmonary arterial hypertension (PAH) is a devastating cardiovascular disease and is a condition that worsens over time, has no treatment, and can eventually lead to a need for lung transplant. Understanding what happens as the condition worsens is therefore critical to understanding how to stop progression. A recent publication in Circulation Research, led by first author Lingli Wang and senior author Marlene Rabinovitch, provided important insights into how to potentially prevent or reverse PAH progression.
PAH means that there is high blood pressure in the blood vessels of the lung, specifically the pulmonary arteries. Smooth muscle cells (SMCs) in the pulmonary arteries (PASMC) contract to move blood. In PAH, these cells lose their contractile apparatus and undergo rapid division and become so numerous that they progressively narrow and occlude the lumen of the pulmonary arteries , increasing the pressure to maintain flow and in so doing cause strain on the heart. A genetic cause of PAH is a mutation in a gene called bone morphogenetic protein receptor, type 2 (BMPR2) and BMPR2 is also reduced in patients with PAH without a mutation.
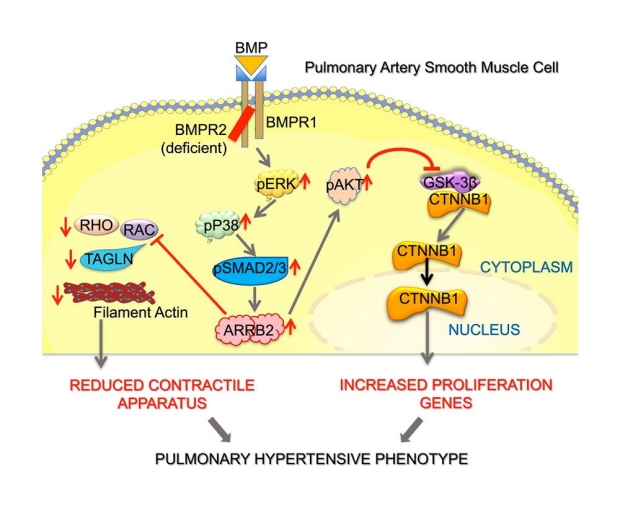
Picture summarizing what was found to happen when BMPR2 is reduced. ARRB2 is increased, which then leads to decreased contractility and increased cell proliferation. Together, that leads to PAH.
The researchers wanted to know whether there was a connection between the loss of BMPR2 and the reduced contractility and heightened multiplication of the PASMC. They used a mouse model lacking Bmpr2 in SMCs combined with cell-based experiments that included cells from patients with BMPR2 mutations. They found that a loss of BMPR2 in PASMCs led to decreased contractility and increased proliferation, which contributes to the characteristic narrowing of the pulmonary arteries. Through a series of sophisticated experiments, the team was able to better understand the steps that occur as a result of reduced BMPR2 in SMC.
These steps include an increase in ARRB2 levels, which then leads to further changes that directly impair contractility and promote cell division or proliferation. Creating this model of what happens as a result of decreased BMPR2 that leads to PAH progression can open the door for considering ways to reverse or slow progression of PAH, something that is critically needed. For example, the authors propose that targeting the elevated ARRB2 could reverse or prevent the increase in PASMCs that leads to the progression of PAH. These results provide a framework with exciting potential for future ways to prevent or reverse PAH progression and uncover the link between loss of BMPR2 and the initiation and worsening of PAH, focusing on the muscle cells of the pulmonary arteries.

Lingli Wang, MD

Marlene Rabinovitch, MD