Reversing Genetic Damage in Pulmonary Arterial Hypertension
by Adrienne Mueller, PhD
April 23, 2021
When Lung Vessels Wither
Blood vessels are our conduits for oxygen and nutrients. Our blood vessels’ inner surface is lined by a single layer of specialized cells called endothelial cells, that have multiple functions including maintaining vascular contractility and permeability and preventing inflammatory cell invasions. When endothelial cells fail in their function, the effect on our system can lead to a host of disorders including atherosclerosis, aneurysms and a devastating disease of the lungs called pulmonary arterial hypertension (PAH). In PAH, endothelial cells fail to control the normal patency and the muscle cells expand and narrow the vessel lumen. Endothelial cells in PAH are also more prone to die, causing the loss of the smallest and most delicate of our blood vessels: microvessels. Those endothelial cells that survive transform in a manner that makes them more like muscle cells and subverts their normal function. Our current treatments for PAH try to dilate the vessels that have narrowed, but there are no existing treatments that reverse the narrowing of the large vessels or regenerate lost microvessels; and for advanced PAH, lung transplantation is the only current cure. To develop better treatments, a clearer understanding of the mechanisms underlying PAH is needed.
Genetic Damage and Genetic Repair
DNA damage and genomic instability go hand in hand with PAH. Failure of DNA repair can eventually lead to cell death and contributes to the loss and narrowing of blood vessels in PAH. It was not known what caused DNA damage or impaired the ability of cells to repair damaged DNA in PAH, but our previous studies led us to believe that this was directly related to a gene, BMPR2, that is mutant or deficient in PAH. One of the roles of BMPR2 is to activate a pair of proteins: PPARγ and p53. Previous studies have shown that both PPARγ and p53 help repair damaged DNA and increasing their activity helps reduce PAH. The question that had not been answered is what genes are co-dependently controlled by these transcription factors that are critical in the ability of the pulmonary arterial endothelial and also muscle cells of the vessel wall to repair damaged DNA, to regenerate lost vessels and to prevent or reverse narrowing of larger arteries in response to an injury that can lead to PAH. A group of scientists in the Rabinovitch lab tackled these questions and their findings were recently reported by first author Jan Hennigs, MD and senior author Marlene Rabinovitch, MD in Circulation Research.
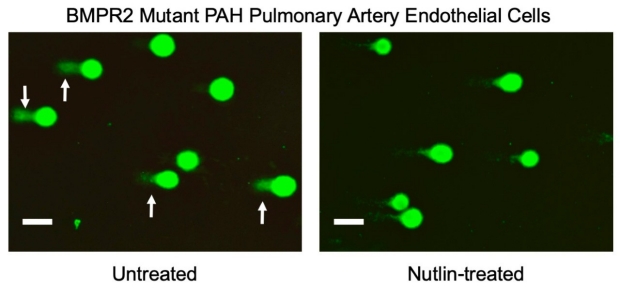
Nutlin Resolves DNA damage by Increasing P53 in PAH Endothelial Cells
The comet tails reveal DNA damage in PAH endothelial cells that is reduced by Nutlin treatment in conjunction with induction of DNA repair genes owing to p53 transcriptional activity.
A New Mechanism of Treatment
Hennigs et al showed that PPARγ and p53 join to form a complex in response to DNA damage. The investigators then demonstrated, through multiple parallel experiments, that loss of BMPR2 signaling reduces the formation of the PPARγ-p53 complex. Hennigs et al went on to identify a host of downstream genes that were regulated by this complex: genes that influence cell survival, regeneration, and DNA repair.
The next step was to figure out how to increase the formation of the PPARγ-p53 complex and bypass the adverse consequences of the reduction in BMPR2. Hennigs et al deployed a pharmacological intervention, Nutlin, to increase p53 levels in a mouse without BMPR2 in endothelial cells, and allow the PPARγ-p53 complex to reform. Once the complex was restored DNA damage was repaired, microvessels were regenerated, and the narrowing of the larger arteries reversed. The investigators have therefore identified a novel strategy for reactivating a molecular genetic repair system that is otherwise not functioning in PAH.
Additional Stanford Cardiovascular Institute-affiliated investigators who contributed to this study include Aiqin Cao, Caiyun (Grace) Li, Minyi Shi, Kazuya Miyagawa, Pin-I Chen, David P. Marciano, Matthew Roughley, Matthew V. Elliott, Rebecca L. Harper, Matthew A. Bill, James Chappell, Jan-Renier Moonen, Isabel Diebold, Lingli Wang, and Michael P. Snyder.

Jan Hennigs, MD

Marlene Rabinovitch, MD