Research

Since the discovery of p53 in 1979, it has become clear that the p53 gene is the most mutated gene in human cancer, with mutations seen in over half of all human cancers, highlighting its fundamental role as a tumor suppressor. Despite this striking observation, it remains unclear which p53-regulated gene networks and which cellular programs facilitate this critical transcription factor’s anti-tumor activity. Understanding these programs is key to understanding how p53 loss promotes deadly cancers like lung and pancreatic cancer which display particularly high rates of p53 mutation. Notably, there are not yet standard-of-care therapies based on targeting the p53 pathway. Although the role of p53 as a sentinel of acute DNA damage is well known, research from our lab has suggested that this activity is not essential for stemming cancer growth. If not the DNA damage response, then which pathways enable the activity of the genome’s most powerful tumor suppressor? Answering this question is fundamental for identifying therapeutic strategies for targeting the p53 pathway. Thus, our laboratory seeks to answer this very question through CRISPR/Cas9 screens, ATAC-sequencing, genetic mouse models, proteomics, single cell transcriptomic, and a wide variety of other techniques available to us through our large network of collaborators at Stanford. Continue reading below for summaries of ongoing projects in our lab, including characterization of the origins of pancreatic cancer, novel p53 target genes, p53’s response to low oxygen, p53’s role in pathology, and more.
Image Credit: Tony Boutelle, Graduate Student at Stanford University
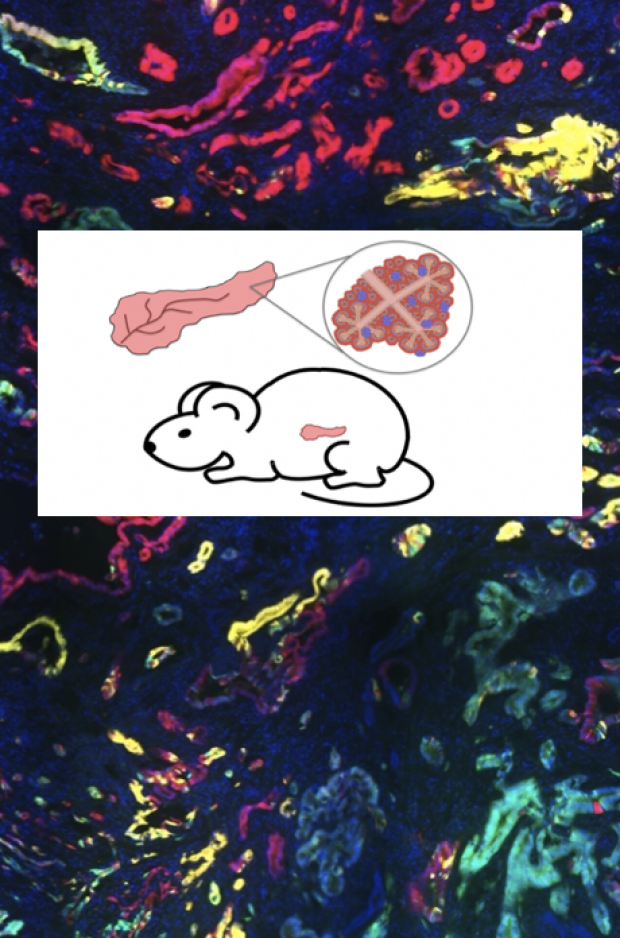
Understanding the origins and evolution of pancreatic cancer
Pancreatic ductal adenocarcinoma (PDAC) is slated to be the 2nd leading cause of cancer deaths by 2026. PDAC, which has a 5-year survival rate of ~10%, is particularly deadly because symptoms manifest late, after the cancer has metastasized, and because treatments are largely ineffective. It is therefore imperative to better understand PDAC initiation and evolution to develop better early detection strategies and therapeutic interventions.
We have used tractable mouse models to activate the Kras oncogene and modulate tumor suppressor genes in either ductal or acinar cells of the pancreas to drive cancer. We have discovered that PDAC can initiate from either cell type, leading to transcriptionally distinct cancers that correlate with specific subtypes of human PDAC. We are deconstructing paths of PDAC development by exploring the transcriptome, chromatin landscape, and tumor microenvironment through single cell RNA-sequencing and spatial transcriptomics. These approaches will elaborate the mechanisms of initiation and paths of tumor evolution in this deadly disease.
Image Credit: Brittany Flowers, Graduate Student at Stanford University

Understanding the “p53 super tumor suppressor”
Which pathways are dysregulated with p53 loss? Understanding the pathways through which p53 acts is critical for understanding how cells change when p53 is inactivated, and, ultimately, for devising therapies to target p53-deficient cells. One approach we have taken to delineate molecular pathways downstream of p53 is to leverage a mutant we generated, known as p5353,54, which is a better tumor suppressor than wild-type p53. Analysis of this mutant revealed Ptpn14, a non-receptor tyrosine phosphatase, as a key tumor suppressor downstream of p53 in pancreatic cancer. Ptpn14 works in part through inhibition of the Yap oncoprotein. We are currently aiming to further understand Ptpn14 through both proteomics and mouse genetic approaches.
Image Credit: Dr. Stephano Spano Mello, Ph.D., Assistant Professor at the University of Rochester

The Mettl3 epitranscriptomic writer amplifies p53 activity
While p53 has been recognized to regulate the epigenome, our recent work has revealed a new facet to p53 function in regulating the epitranscriptome. By characterizing the p53-interacting proteome, we identified Mettl3 as an RNA methyltransferase that interacts with p53. We find that Mettl3 amplifies p53 function in tumor suppression by helping to stabilize p53 and to promote modification of downstream p53 target gene transcripts to enhance their expression. We are currently exploring these mechanisms in more detail. Moreover, this new aspect of regulation may be critical for p53 to drive cell state changes, an idea we are testing currently.
Image Credit: Richard Zhao, Attardi Lab Member at Stanford University
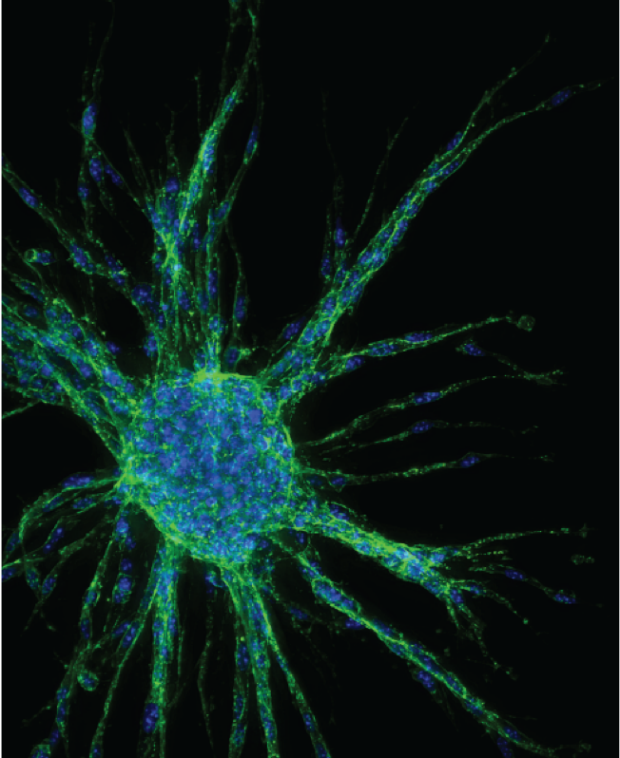
Pleiotropic effects of p53 loss revealed by studies in physiological oxygen
Which cellular functions account for p53-mediated tumor suppression? Our recent studies reveal that p53 loss drives numerous changes in cellular biology, including phenotypes related to survival in response to stress, DNA repair, migration, invasion, and metabolism. The pleiotropic effects seen upon p53 inactivation help to explain its frequent loss in cancer. Notably, some of these phenotypes were only revealed through studies in physiological oxygen, underscoring the importance of using physiologically relevant culture conditions. How does oxygen affect p53 status? How does the effect of p53 on cell biology change depending on oxygen levels? How does the proclivity of a cell to lose p53 depend on oxygen levels? How does p53 biochemical function change at different oxygen levels? These remain key questions and will help us understand tumor suppressor signaling in vivo.
Image Credit: Dr. Liz Valente, Ph.D., Research Associate at Heligenics

Deconstructing molecular programs of p53-mediated tumor suppression.
Which pathways are dysregulated with p53 loss? Understanding the pathways through which p53 acts is critical for understanding how cells change when p53 is inactivated, and, ultimately, for devising therapies to target p53-deficient cells. We have used a variety of approaches to delineate molecular pathways downstream of p53. In one approach, shRNA and CRISPR/Cas9 screens were used to identify p53 target genes critical for tumor suppression. These analyses converged on the p53 target gene, Zmat3, which shows broad tumor suppressor activity, in leukemia, lung adenocarcinoma, and liver cancer. Integrative CLIP-seq and RNA-seq analyses revealed Zmat3 as a key regulator of alternative splicing, illuminating a splicing program in the p53 tumor suppression response. We are currently using CRISPR/Cas9 screens to identify Zmat3 cooperating partners in tumor suppression. These approaches will allow us to fully deconstruct the networks underlying p53-mediated tumor suppression.
Image Credit: Kathryn Bieging-Rolett, Ph.D., Scientist II at HiberCell

p53 induces a constellation of developmental defects in the absence of apoptosis
Not only does p53 loss promote cancer development but p53 hyperactivation also drives a range of pathologies, in developmental syndromes, aging, and neurodegenerative diseases. These observations underscore the importance of balanced p53 activity and shape our thinking of how to best develop cancer therapies based on modulating the p53 pathway. Interestingly, we showed that p53 hyperactivation during embryogenesis causes a constellation of very specific developmental phenotypes typical of a human syndrome known as CHARGE (coloboma, heart defects, atresia of the choanae, retarded growth and development, genital hypoplasia, and ear abnormalities). How p53 triggers such specific abnormalities, and how it promotes distinct sets of developmental defects in different syndromes has been enigmatic. We have addressed this question by modulating the spatial expression pattern, intensity, and timing of p53 activation during mouse development. These studies have helped to decode the basis of p53 driven developmental defects, and we are now working to understand the specific molecular pathways through which p53 acts. Interestingly, although apoptosis is associated with p53 action, it is not essential for the manifestation of the p53-driven developmental defects. We are currently exploring whether other cell death pathways or responses underlie these detrimental phenotypes. Understanding the pathways of p53 action is critical for devising the best strategies to target p53 in pathological contexts.
Image Credit: Dr. Margot Bowen, Ph.D., Support Scientist at 10X Genomics