Learn more about funded research in the Oro Lab

Oro lab and EB iPS Consortium Receive $5M CIRM Translation Award
Our lab and the EB iPS Consortium, composed of Stanford's Marius Wernig, Univ Colorado Dennis Roop, and Columbia's Angela Christiano, will work with the newly formed Stanford Center for Definitive and Curative Medicine and the Laboratory for Cell and Gene Medicine to develop a clinically robust and widely available treatment. We hope our efforts will also facilitate other normal and genetically corrected tissue replacement therapies that the center aims to develop at Stanford.

Stanford announces new Center for Definitive and Curative Medicine July 2017
To help accelerate development of the iPS-derived tissue regeneration product in our lab, Tony helped to co-found the Center for Definitve and Curative Medicine with Maria Grazia Roncarolo and Matthew Porteus--"one dose is a cure". Stem cell and gene therapy hold enormous promise to cure conditions with well-defined genetic causes by engineering cells to treat disease or altering a patient’s personal DNA to “fix” an abnormality. To bring these new stem cell and gene therapies to their patients, Stanford Medicine has announced the opening of the Stanford Center for Definitive and Curative Medicine, a joint initiative of the Stanford University School of Medicine, Stanford Health Care and Stanford Children’s Health.
The center provides the organizational and physical infrastructure to support investigator-initiated clinical translational studies on stem cell and gene therapy from initial discovery through completion of clinical proof-of-concept studies. Stanford Medicine is in a unique position to develop the CDCM because of its outstanding expertise in disease pathophysiology, cell and stem cell biology, and an optimal and collaborative environment between the medical school and the hospitals.

Stanford cGMP Cell Therapy Facility opens September 2016
Epidermolysis bullosa patients suffer profound skin fragility, ineffective wound healing and persistent erosions leading to chronic wounding and longer-term complications of scarring and increased risk of malignancy. Traditional medical approaches have focused on either a surgical approach, where the diseased tissue is removed, or a medical approach, where a pill is provided to the patient. EB and diseases like EB fall outside the realm of traditional medical approach and require a third type of approach that focuses on tissue regeneration through the correction of the diseased protein. The Stanford EB Consortium represents a campus-wide collaboration of clinicians and scientists focused on developing this third tissue approach using novel molecular and cellular therapies to alleviate patient suffering. Accelerating this effort is the construction of a cGMP Cell Therapy Facilty on the Stanford campus through a joint effort of Stanford Hospital, Packard Hospital and Stanford medical school. This facility will allow the development of novel cell-based treatments and teach a new generation of physicians and scientists about definitive genetic therapy.

Therapeutic Reprogramming for Epidermolysis Bullosa using AAV-DJ-mediated correction
Patients with recessive dystrophic epidermolysis bullosa (RDEB) lack functional type VII collagen and suffer severe blistering and chronic wounds that ultimately lead to infection and development of lethal squamous cell carcinoma. The discovery of induced pluripotent stem cells (iPSCs) and the ability to edit the genome bring the possibility to provide definitive genetic therapy through corrected autologous tissues. Our lab has developed a scalable and cGMP compatible protocol to generate patient-derived COL7A1 gene-corrected epithelial keratinocyte sheets for autologous grafting. iPSC-derived keratinocytes are produced with minimal heterogeneity and secreted wild-type type VII collagen, resulting in stratified epidermis in vitro and in vivo in mice. Sequencing of corrected cell lines prior to tissue formation revealed heterogeneity of cancer-predisposing mutations, allowing us to select COL7A1 corrected banks with minimal mutational burden for downstream epidermis production.
Highly Recombinogenic AAV for Genome Editing of Somatic and ES Cells
An explosion in the ability to edit the human genome, in conjunction with the identification of human tissue and embryonic stem cells allows for the first time in history the ability for “definitive genetic therapy” through the correction of genetic mutations in stem cells. We investigated the efficiency of genome editing tools with an eye for scalability and cGMP compatibility. We have explored conventional targeting, zinc finger nucleases, transcriptional activator-like effector nucleases (TALENs), and CRISPR/Cas9-based editing. In addition, in collaboration with Mark Kay at Stanford, we have found a novel adeno-associated viral AAV variant (AAV-DJ) that has high recombinogenic activity. AAV-DJ–mediated targeting (AT) has the advantage that AAVs have previously been used in human gene therapy trials, have low off-target rates, and are easily detectable. In our initial paper, we demonstrated the ability of AAV-DJ to directly correct the LamA3 mutation in Junctional Epidermolysis Bullosa patient keratinocytes. This provided proof-of-principle that direct correction, without the use of selectable markers, could be used clinically. Moreover, in a followup paper, we showed that an AAV-DJ variant could correct the COL7A1 locus in Dystrophic Epidermolysis bullosa induced –pleuripotent cells, demonstrating its usefulness in manufacturing protocols for the Therapeutic Reprogramming of skin. Because AAV-DJ–mediated homologous recombination rates were comparable to those of CRISPR/Cas9, and potentially safer and easier to deliver, we concluded that AAV-DJ–mediated genome editing would provide the ideal genome-editing system for correction during therapeutic iPSC manufacturing.
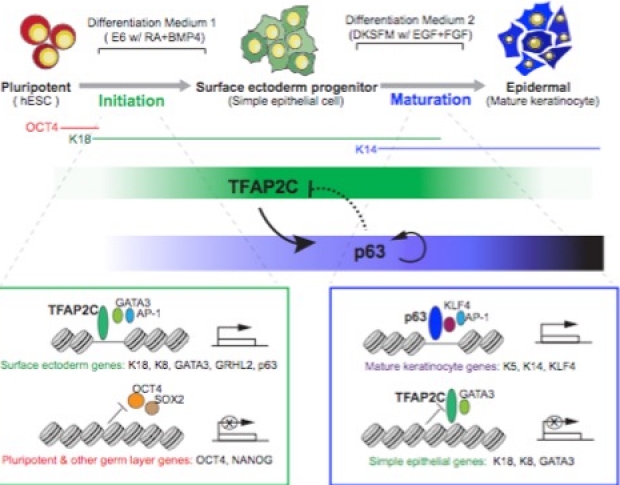
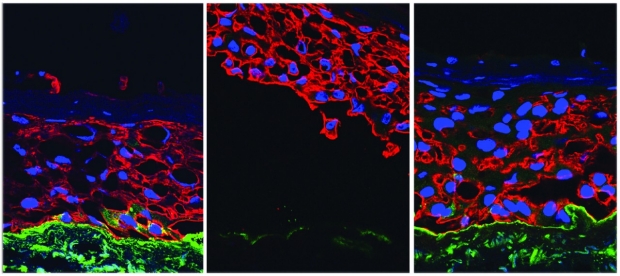
New transcription factor inference model reveals initiation and maturation networks for stratified epidermal differentiation
Tissue development results from lineage-specific transcription factors (TF) programming a dynamic chromatin landscape through progressive cell fate transitions. We interrogate the epigenomic landscape during epidermal differentiation and, in collaboration with Wing Wong's lab, create an inference network that ranks the coordinate effects of TF-accessible regulatory element-target gene expression triplets on lineage commitment. We discover two critical transition periods: surface ectoderm initiation and keratinocyte maturation, and identify TFAP2C and p63 as lineage initiation and maturation factors, respectively. Surprisingly, we find that TFAP2C, and not p63, is sufficient to initiate surface ectoderm differentiation, with TFAP2C-initiated progenitor cells capable of maturing into functional keratinocytes. Mechanistically, TFAP2C primes the surface ectoderm chromatin landscape and induces p63 expression and binding sites, thus allowing maturation factor p63 to positively auto-regulate its expression and close a subset of the TFAP2C-initiated early program. Our work provides a general framework to infer TF networks controlling chromatin transitions that will facilitate future regenerative medicine advances.

Genomic analysis of Human Skin Differentiation using our novel in vitro differentiation protocol
In the course of developing the Therapeutic Reprogramming protocol for skin, we developed a reliable in vitro differentiation protocol for skin. This has allowed us to take a intensive genomics and bioinformatic / network analysis approach to understand how human skin progresses through pluripotent, simple and stratified epithelium in response to developmental morphogen retinoic acid and BMP. Our studies using cohesin Hi-ChIP technology to analyze chromatin conformation have shown that developmental morphogens work with major lineage selectors like p63 to properly fold local chromatin and connect key regulatory elements to their neighboring genes.
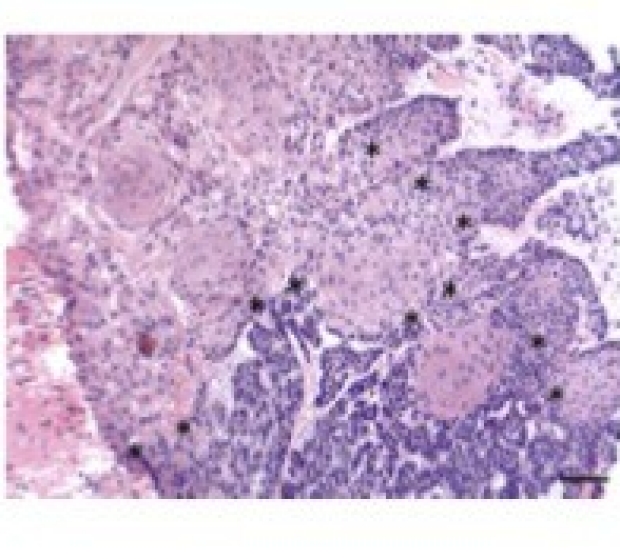
Genetic Mutations underlying phenotypic plasticity in basosquamous carcinoma
Basosquamous carcinoma (BSC) is an aggressive skin neoplasm with the features of both basal cell carcinoma (BCC) and squamous cell carcinoma (SCC). We sought to characterize the genomic alterations underlying sporadic BSC to elucidate the derivation of these mixed tumors.45% of the BSCs harbor recurrent mutations in the SWI/SNF complex gene, ARID1A, and evolutionary analysis revealed that ARID1A mutations occur after PTCH1 but before SCC driver mutations, indicating that ARID1A mutations may bestow plasticity enabling squamatization. Overall, these results support the genetic derivation of BSCs from BCCs and highlight potential factors involved in modulating tumor reprogramming between basaloid and squamatized phenotypes.

Loss of Primary Cilia Drives Switching from Hedgehog to Ras/MAPK Pathway Dependence in Resistant BCCs
We previously observed epigenetic pathway switching from BCCs to a different subtype of skin cancer, SCC as a mechanism of tumor evolution and resistance. In this work, we identify the primary cilium as a critical determinant controlling tumor pathway switching. Smoothened inhibitor-resistant BCCs have an increased mutational load in ciliome genes, resulting in reduced primary cilia and HH pathway activation compared with naive or Gorlin syndrome patient BCCs. Gene set enrichment analysis of resistant BCCs with a low HH pathway signature showed increased Ras/MAPK pathway activation. Our results provide insights into BCC treatment and identify the primary cilium as an important lineage gatekeeper, preventing HH-to-Ras/MAPK pathway switching.

aPKC and HDAC1 regulate GLI1 activity by controlling LAP2-dependent movement from nuclear lamina to chromatin
Understanding transcription factor navigation through the nucleus remains critical for developing targeted therapeutics. The GLI1 transcription factor must maintain maximal Hedgehog pathway output in basal cell carcinomas (BCCs), and we have previously shown that resistant BCCs increase GLI1 deacetylation through atypical protein kinase Ci/l (aPKC) and HDAC1. Here we identify a Lamin-Associated Polypeptide 2 (LAP2) isoform-dependent nuclear chaperoning system that regulates GLI1 movement between the nuclear lamina and nucleoplasm to achieve maximal activation. LAP2β forms a two-site interaction with the GLI1 zinc-finger domain and acetylation site, stabilizing an acetylation-dependent reserve on the inner nuclear membrane (INM). By contrast, the nucleoplasmic LAP2α competes with LAP2β for GLI1 while scaffolding HDAC1 to deacetylate the secondary binding site. aPKC functions to promote GLI1 association with LAP2α, promoting egress off the INM. GLI1 intranuclear trafficking by LAP2 isoforms represents a powerful signal amplifier in BCCs with implications for zinc-finger based signal transduction and therapeutics.

Cytoskeletal/Fibrotic signaling through SRF-MRTF confers Drug resistance
Using multi-dimensional genomic analysis of animal models and our patients with drug-resistant BCCs, we have found that Serum Response Factor/Myocardin Related Transcription Factor (SRF/MRTF) activation is a common and powerful mechanism of drug resistance. SRF/MRTF have previosly been associated with fibrosis and inflammation, and MRTF inhibitors significantly slowed the growth of drug-resistant basal cell carcinomas in mice and signaling in primary patient tumors, highlighting the therapeutic potential of MRTF-class of drugs.
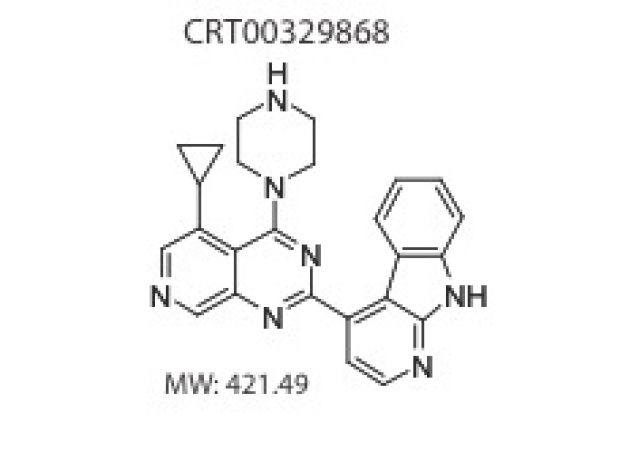
Combined inhibition of atypical PKC and histone deacetylase 1 act synergistically in the treatment of BCC
We examined patients with Smo inhibitor resistant BCCs and proteins that interact with the primary cilium to demonstrate that the polarity kinase atypical protein kinase C iota/lamda (aPKC) is critical for Hh-dependent processes. Using a drug repositioning screen we show that aPKC acts in the nucleus directly on the Gli transcription factor complex, acting in concert with HDAC1 to promote chromatin association and tumor resistance. Our work implicates the kinase as a new, tumor-selective therapeutic target for the treatment of Smo-inhibitor resistant cancers and led us to identify the first promising candidate aPKC inhibitor for BCCs.
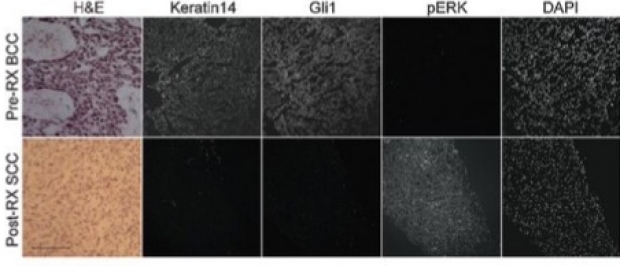
Pathway Switching from BCC to SCC during tumor resistance
Our group found a novel mechanism that advanced BCCs frequently acquire resistance to Smoothened (SMO) inhibitors: through tumor type / signaling pathway switching from BCCs to squamous cell carcinomas (SCC). Both BCC and SCC derive from a common cancer stem cell, with hedgehog pathway driving BCCs and RAS/MAPK signaling driving SCC. We had previously observed SCCs arising out of the same location where a drug-sensitive BCC had been treated. Our group showed that the SCC was related by lineage to the BCC but had shut off the hedgehog pathway and turned on RAS/MAPK. This work provides the molecular underpinnings for tumor type pathway switching. Please see related work from our Stanford colleagues Ransohoff et al. NEJM 2015 373:1079-82.
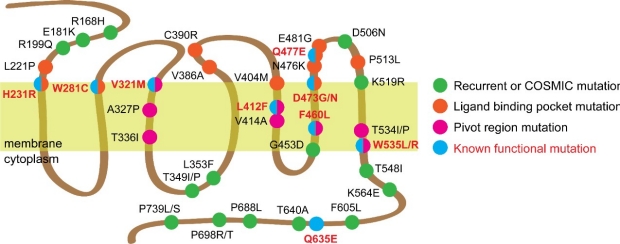
Tumors "Roll the Genetic Dice" to generate drug resistance
Genomic analysis by our group and others have revealed that BCCs are typically diploid and carry a high frequency of non-silent single nucleotide variants (SNVs) compared to other cutaneous and non-cutaneous tumors. Given their high mutational load, how these variants confer selective tumor growth without deleterious effects remains poorly understood. We previously identified and functionally validated nine SMO mutations that drive the majority of drug resistance in BCC through two distinct mechanisms that maintain HH signaling in the presence of drug: induction of constitutive activity or disruption of ligand binding (see below). However, SMO mutations with unclear function are frequently found across many HH and non-HH dependent cancers with drug-resistant BCCs bearing the highest rate of recurrent mutations at 66%. Using a sensitive functional assay, our results reveal a surprising frequency of neutral and inactivating SMO variants in our drug-resistant BCC tumor population. Our data supports a model where tumors have an inherent advantage to generate as many genetically diverse clones as possible to cultivate a competitive environment that forces clonal competition as a way to grow. This ability to “roll the genetic dice” creates many mutations in key genes like SMO that would have activating, neutral, or negative effects on the cell. However, a small percentage of clones fortunate enough to contain activating mutations would continue to divide and contribute to a larger fraction of the tumor mass.
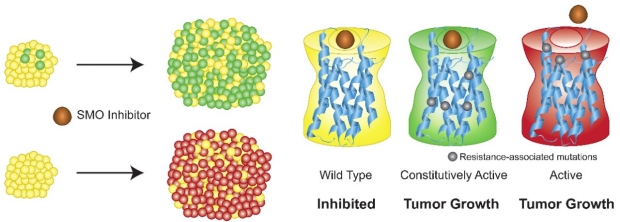
Fitness of Smoothened Variants in Drug Resistant human BCCs
Advanced basal cell carcinomas (BCCs) frequently acquire resistance to Smoothened (SMO) inhibitors through unknown mechanisms, providing a unique opportunity to study human tumor evolution in patients through whole exome, RNA, and targeted sequencing. We find SMO mutations in 50% of resistant BCCs compared with 6% of untreated BCCs. Alterations both ligand binding pocket mutations that define sites of inhibitor binding constitutively active SMO mutants define pivotal residues of SMO that ensure receptor autoinhibition. Both classes of SMO variants respond to the aPKC-i/l inhibitor PSI and GLI2 antagonist ATO that operate downstream of SMO, setting the stage for the clinical use of GLI antagonists we are developing in our lab.
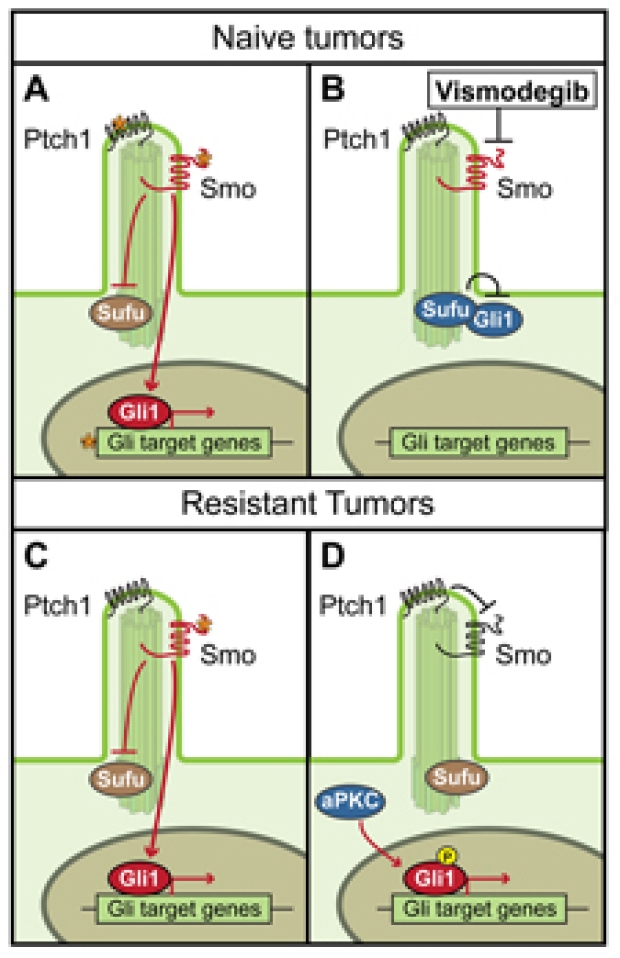
Mechanisms of Tumor Evolution and Drug Resistance in BCCs
Developing effective targeted therapies to dispatch tumors before they evolve resistance requires knowledge of available escape pathways. This is especially critical given that resistant clones are likely present in small numbers at the time of treatment initiation. Given the pathway and compensatory alterations in Hh-dependent tumors thus far, dual targeting of the most downstream component of the pathway, and the compensatory pathway(s), will likely generate optimal therapies. Gli transcription factors ultimately transduce the signal from Hh ligand; moreover, escape pathways that bypass Smo still activate Gli. Targeting Gli directly or the signaling components that activate Gli could prove quite successful as the next level of therapy. We are excited about developing aPKC inhibitors to treat hedgehog-dependent tumors.
The blueprint to develop the first Hh pathway inhibitors came from impressive efforts from the Hh and cancer communities, beginning with identification of Hh pathway components and their roles in cancer and continuing with intense screening and medicinal chemistry that refined drug targets for optimal human use. The challenge for the future is to better understand common pathway-dependent genetic and compensatory escape pathways that evolve from clonal populations within tumors and design combination therapies to block them before further evolution takes place. Through improved genomics, cell biology, and medicinal chemistry, this may be a race medical research can win.
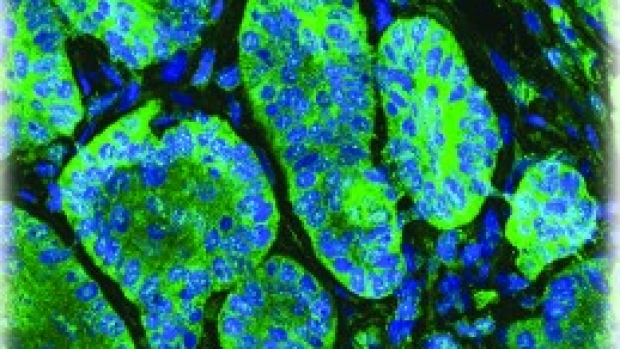
Atypical Protein Kinase C in Gli Activation
Basal cell carcinoma (BCC) growth requires high levels of Hedgehog (Hh) signaling through the transcription factor Gli. After the initial discovery of the connection between Hh signaling and BCCs, the Stanford BCC Consortium worked to test the first Hedgehog pathway inhibitors. While inhibitors of membrane protein Smoothened (Smo) effectively suppress Hh signaling, early tumor resistance illustrates the need for additional downstream targets for therapy. In collaboration with the Stanford Consortium, we have sequenced RNA and DNA from sensitive and drug resistance tumors to better understand how tumors evolve. One of the initial observations was the dramatic increase in the levels and activity of the polarity kinase atypical Protein Kinase C iota/lambda (aPKC) and its role as a novel Gli activator. aPKC and its polarity signaling partners colocalize at the centrosome and form a complex with Missing-in-Metastasis (MIM), a scaffolding protein that potentiates Hh signaling. Genetic or pharmacological loss of aPKC function blocks Hh signaling and proliferation of BCC cells. aPKC is a Hh target gene that forms a positive feedback loop with Gli and exhibits elevated levels in BCCs. Genome-wide transcriptional profiling shows that aPKC and Smo control the expression of similar genes in tumor cells. aPKC functions downstream of Smo to phosphorylate and activate Gli1, resulting in maximal DNA binding and transcriptional activation. Activated aPKC is upregulated in Smo-inhibitor resistant tumors and targeting aPKC suppresses signaling and growth of resistant BCC cell lines. Our results demonstrate aPKC is critical for Hh-dependent processes and implicates aPKC as a new, tumor-selective therapeutic target for the treatment of Smo-inhibitor resistant cancers.
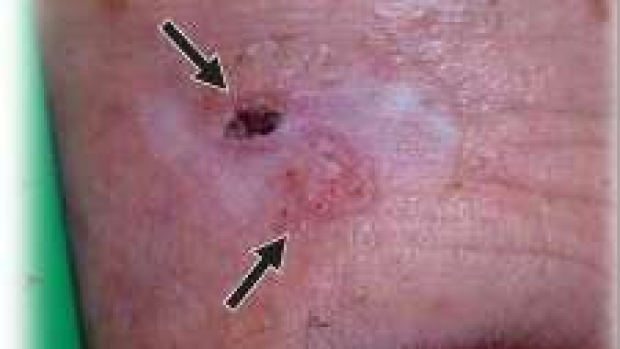
Acquired Resistance within 1 year in 20% of Smo Inhibitor treated patients
Previous observation with advanced BCC patients showed that 55-70% of patients had primary resistance to the Smoothened inhibitor vismodegib (Sekulic et al. 2012). Secondary (acquired) resistance is characterized by regrowth of a tumor after initial shrinkage. In this study we show that of the patients that initially respond, approximately 20% will develop secondary resistance with a median time to regrowth of about 1 year. This reiterates the need for combination therapy and frequent skin examinations for those with advanced BCC.
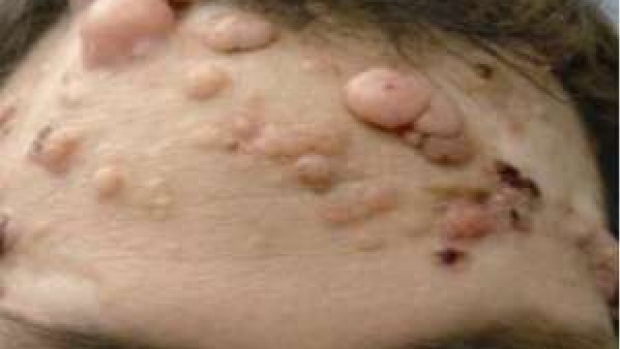
Efficacy of First Hedgehog Pathway Inhibitor Targeting Smoothened
In this multicenter, international, two-cohort, nonrandomized study funded by Genentech, Stanford patients with metastatic basal-cell carcinoma or those with locally advanced basal cell carcinoma who had inoperable disease, received the first hedgehog pathway inhibitor. The study demonstrated that Vismodegib is effective in 30-45% of patients, and validated Smoothened as a therapeutic target. In the same issue of the New England Journal of Medicine, we describe a novel patient in the trial with advanced BCC from a (7;Y) translocation driving Sonic Hedgehog in the skin.
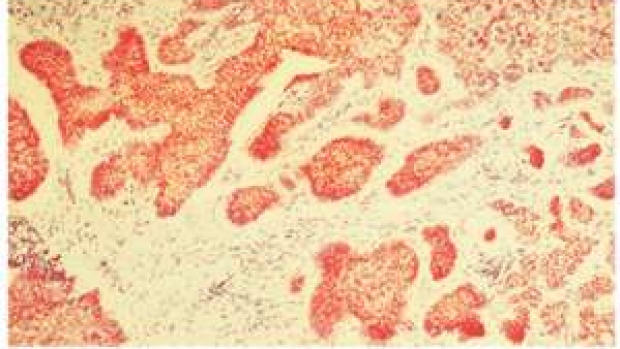
Basal Cell Carcinomas in Mice Overexpressing Sonic Hedgehog Ligand
The Scott lab found that mutations in the PTCH1 tumor suppressor cause Basal cell nevus syndrome (Gorlin's), a disorder where patients develop hundreds of BCCs and other hedgehog-dependent cancers. Gene regulatory relationships defined in the fruit fly suggest that SHH overproduction should mimic PTCH1 loss if SHH is sufficient to drive tumorigenesis. We show that SHH overexpression is sufficient to induce BCCs in mice and reinforce the unique dependence of BCCs on the hedgehog pathway for growth. We went on, in collaboration with Paul Khavari, to show a similar finding in human skin using SHH-retroviral infected, regenerated human epidermis. Fan, Oro, Scott and Khavari Nat Med 3:788-92 1997.
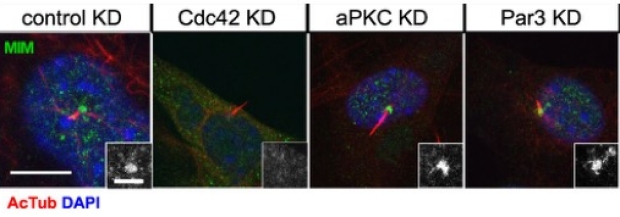
Actin Polymerization controls cilia-mediated signaling
Primary cilia are polarized organelles that allow detection of extracellular signals such as Hedgehog (Hh). How the cytoskeleton supporting the cilium generates and maintains a structure that finely tunes cellular response remains unclear. Here, we find that regulation of actin polymerization controls primary cilia and Hh signaling. Disrupting actin polymerization, or knockdown of N-WASp/Arp3, increases ciliation frequency, axoneme length, and Hh signaling. Cdc42, a potent actin regulator, recruits both atypical protein pinase C iota/lambda (aPKC) and Missing-in-Metastasis (MIM) to the basal body to maintain actin polymerization and restrict axoneme length. Transcriptome analysis implicates the Src pathway as a major aPKC effector. aPKC promotes whereas MIM antagonizes Src activity to maintain proper levels of primary cilia, actin polymerization, and Hh signaling. Hh pathway activation requires Smoothened-, Gli-, and Gli1-specific activation by aPKC. Surprisingly, longer axonemes can amplify Hh signaling, except when aPKC is disrupted, reinforcing the importance of the Cdc42–aPKC–Gli axis in actin-dependent regulation of primary cilia signaling.
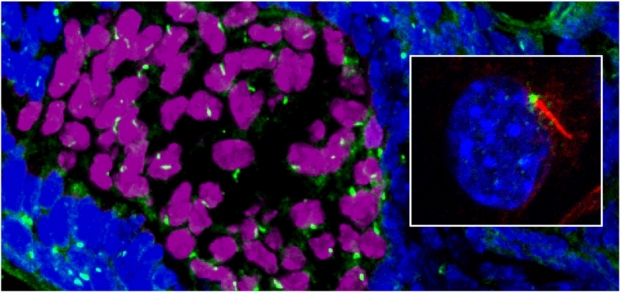
Mtss1 / Src Kinase Signaling Cassette Regulates Ciliogenesis
The signal strength of development morphogens and oncogenes plays a critical role in both morphogenic patterning and tumor growth and resistance. An emerging mediator of signal reception is the microtubule-based organelle, the primary cilium. The small 2 mm sensory structure provides directional signal specificity by concentrating receptor subtypes and downstream signaling cascades into a highly polarized structure. The primary cilium provides critical regulation of the Hedgehog pathway, regulating the interactions of Patched, Smoothened, and Gli in a ligand-dependent manner. We have identified a novel Shh-induced cytoskeletal gene Missing-in-Metastasis (MTSS1 or MIM) and have shown MTSS1 is required for Shh signaling in BCC cells. MTSS1 localizes to the basal body of the primary cilium and regulates vesicle movement and ciliary length by antagonizes the activity of Src-family kinases and actin regulators like Cortactin. MTSS1 overexpression can increase Gli1 activity. We have identified MTSS1-interacting proteins and found that the polarity pathway including Atypical Protein Kinase C, also localizes to the basal body, and are currently investigating the role of aPKC and other MTSS1-interacting proteins in ciliary function and tumor progression.
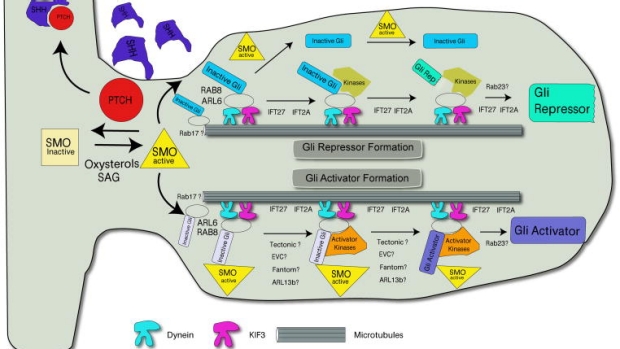
Primary cilium and the regulation of hedgehog signaling
Intense focus has been centered around how the primary cilia transduces the hedgehog signal from smoothened to the Gli transcription factors. New data indicate that ligand and signaling lipids help regulate small GTPase-dependent accumulation and activity of signaling components. These component regulate vesicle transport, regulatory post-translational modifications, and signaling intensity.
MTSS1/Src family kinase dysregulation underlies multiple inherited ataxias
The genetically heterogeneous spinocerebellar ataxias (SCAs) are caused by Purkinje neuron dysfunction and degeneration, but their underlying pathological mechanisms remain elusive. The Src family of nonreceptor tyrosine kinases (SFK) are essential for nervous system homeostasis and are increasingly implicated in degenerative disease. Here we reveal that the SFK suppressor Missing-in-metastasis (MTSS1) is an ataxia locus that links multiple SCAs. MTSS1 loss results in increased SFK activity, reduced Purkinje neuron arborization, and low basal firing rates, followed by cell death. Surprisingly, mouse models for SCA1, SCA2, and SCA5 show elevated SFK activity, with SCA1 and SCA2 displaying dramatically reduced MTSS1 protein levels through reduced gene expression and protein translation, respec- tively. Treatment of each SCA model with a clinically approved Src inhibitor corrects Purkinje neuron basal firing and delays ataxia progression in MTSS1 mutants. Our results identify a common SCA therapeutic target and demonstrate a key role for MTSS1/SFK in Purkinje neuron survival and ataxia progression.

Mtss1, a founding member of the I-BAR family of cytoskeletal regulators, prevents human metastasis
The actin cytoskeleton is essential for cell viability and plays critical roles in cell signaling, proliferation, motility, and survival. Normal physiological functions are associated with a diversity of actin cytoskeletal regulatory proteins and their functions. However, it is becoming clear that local, rather than global, actin cytoskeletal regulation plays critical roles in homeostatic signaling and cancer progression. A major challenge in cell biology is to understand key mechanisms of actin regulation and the range of functions that they control in order to develop therapeutic targets and better understand the clinical context in which to use them. One family of context-dependent cytoskeletal regulators is the I-BAR (Bin/amphiphysin/Rvs) family of cytoskeletal regulators. Our lab focuses on the in vitro and in vivo functions of one of the founding members Missing-in- Metastasis (MIM/Mtss1) in homeostasis and neoplasia. Mtss1 has been identified as a central inhibitor of metastasis in cohorts of human melanoma, breast cancer, and urothelial cancers. Our lab studies how Mtss1 contributes to the metastatic phenotype and how we can develop novel therapeutics for patients with metastatic tumors.
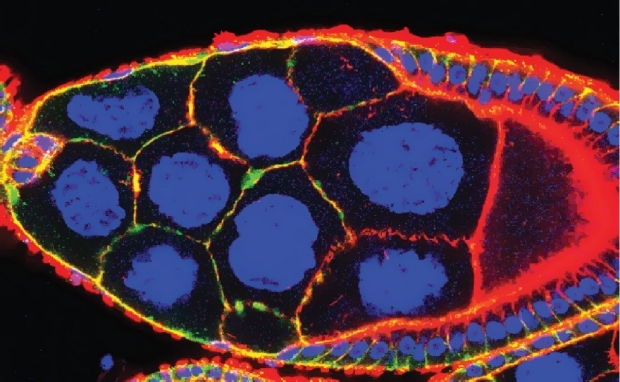
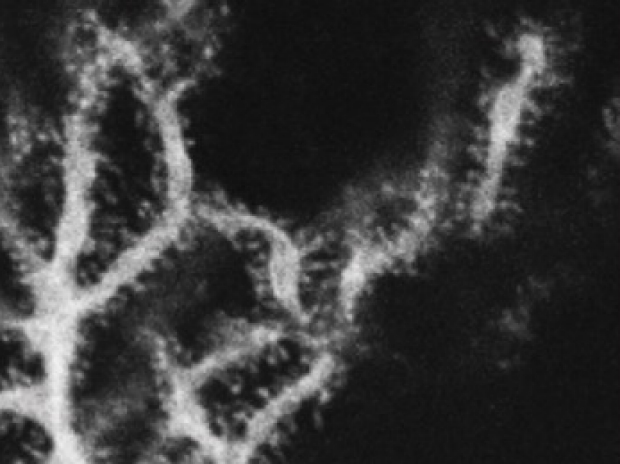
Fly MTSS1 regulates directional migration through Src Kinase mediated endocytosis
Although directed cellular migration facilitates the coordinated movement of cells during development and repair, the mechanisms regulating such migration remain poorly understood. MTSS1 is a defining member of the inverse Bin/Amphiphysin/Rvs domain (I-BAR) subfamily of lipid binding, cytoskeletal regulators whose levels are altered in a number of cancers. We provide the first genetic evidence that an I-BAR protein regulates directed cell migration in vivo. Drosophila MIM (MTSS1) (dmim) is involved in Drosophila border cell migration (see normal migration left below marked with moesin-GFP), with loss of dmim function resulting in a lack of directional movement by the border cell cluster (right, below). In vivo endocytosis assays combined with genetic analyses demonstrate that the dmim product regulates directed cell movement by inhibiting endocytosis and antagonizing the activities of the Src kinase/ CD2- associated protein/cortactin complex in these cells. These studies demonstrate that DMIM antagonizes pro-endocytic components to facilitate polarity and localized guidance cue sensing during directional cell migration.
wild type
MTSS1 mutant Border Cells