Research
Brief Overview of Ongoing Projects
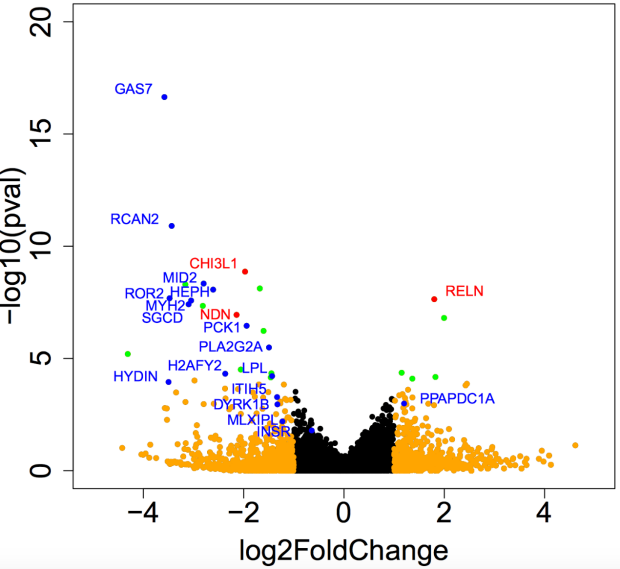
High-Throughput Molecular Profiling
We are using several established or emerging high-throughput, genome-wide techniques such as RNA-seq, HiChIP and ATAC-seq to study expression patterns, transcription factor binding and chromatin interactions and accessibility. Applying these methods in cells of high relevance to insulin resistance, such as adipocytes, hepatocytes and skeletal myocytes, can improve our ability to disentangle genetics of insulin resistance and related traits.
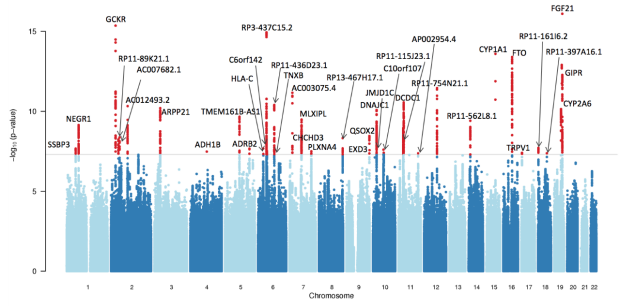
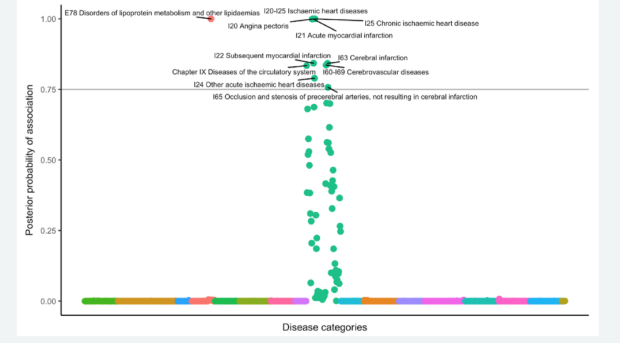
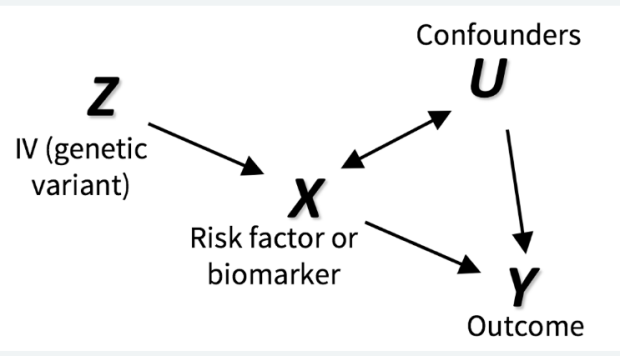
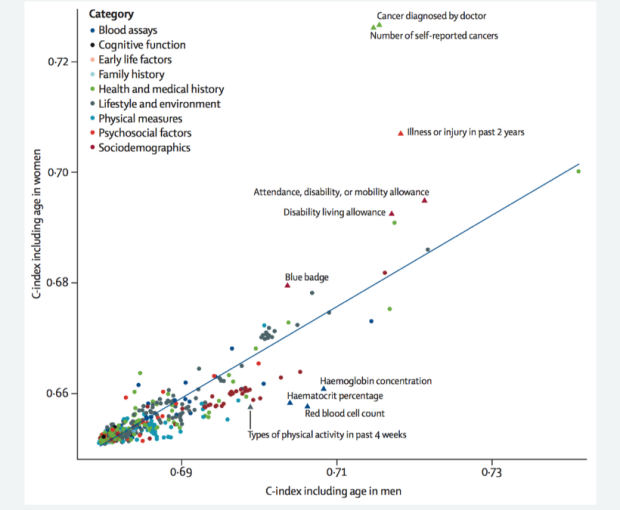
In some projects, we use colocalization methods to systematically prioritize the most likely causal gene in loci associated with insulin resistance and related traits (such as fasting insulin, glucose, waist-hip ratio, HDL cholesterol and triglycerides). These methods seek to determine whether the same variants in a locus are responsible for signals from diwerent layers of high-throughput molecular data, oxen GWAS and eQTL signals. We can also integrate this with other layers of highthroughput molecular data, such as ATAC-seq - to study chromatin accessibility - and HiChIP - a novel method for sensitive and ewicient analysis of protein-centric chromosome conformation.

Pooled CRISPRi Screens
We have identified a large number of loci associated with insulin resistance and related traits using human genetics. By use of pooled genetic screens based on CRISPR, which enable hundreds to thousands of programmed perturbations per experiment, we can prioritize the most likely candidate genes within these loci using unbiased experimental approaches.
We are currently working with two diwerent strategies for phenotypic readouts on the pools of genome-engineered cells. In one project, we apply single-cell RNA sequencing using the recently updated CROP-seq protocol as the readout axer CRISPRi pooled gene perturbation. Another approach is based on reporter gene expression changes following pooled sgRNA delivery for CRISPR-mediated perturbations. The pooled screening approach allows us to evaluate many genes in parallel in a relevant biological context, and the single cell RNA sequencing helps us dissecting the mechanisms underlying changes in insulin resistance and related phenotypes.
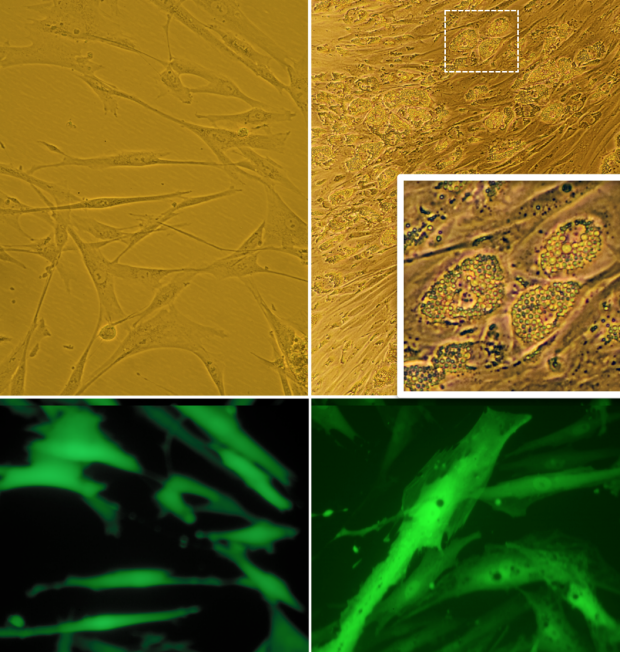
In Vitro Studies in Adipocytes and Other Cell Types
To functionally characterize genes putatively implicated in development of insulin resistance, we work with CRISPR gene perturbations in adipocytes, hepatocytes and skeletal myocytes followed by functional assays relevant to insulin resistance. We primarily work with human cell lines including SGBS preadipocytes, HepG2 hepatocytes and HMCL-7304 skeletal myocytes; but for some applications, we also use murine cell lines, such as 3T3-L1 or OP9 adipocytes, AML12 hepatocytes and C2C12 myocytes, as well as primary cells.
We employ CRISPRi and CRISPRa approaches perturbing target gene expression to investigate cellular phenotypes. In addition, CRISPRi can be used to ewectively, specifically, and homogeneously silence expression of up to 2-3 genes simultaneously, which also allows us to test for epistasis among target genes. Axer specific target gene perturbation, we assess the ewect of gene regulation on a series of phenotypes awected by insulin resistance; specifically, basal and insulin-stimulated glucose uptake, lipolysis, insulin signaling, adipogenesis (adipocytes only), mitochondrial function, fatty acid oxidation, metabolomics and RNA.
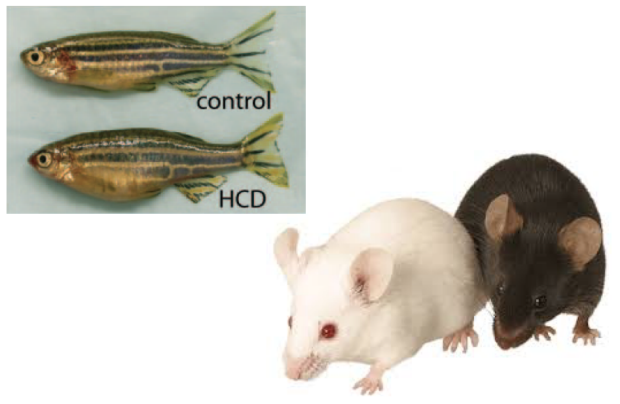
In Vivo Studies
For selected genes showing insulin resistance phenotypes in human genetics and in vitro experiments, we proceed to in vivo models to study physiological ewects axer gene perturbation. Over the past years, we have participated in the development of zebrafish models to allow quantification of body size, lipid accumulation in liver and body fat, pancreatic ß-cell number, liver size and vascular accumulation of lipids. More recently, we are focusing more on mouse models which have proven to be critical in the assessment of insulin resistance, recapitulating expected phenotypes for Mendelian genetic forms of insulin resistance.
Our mouse experiments include feeding on standard chow or high-fat diet followed by tracking of body weight, assessing fasting lipids and glucose, as well as performing insulin and glucose tolerance testing. In selected projects, we measure whole-body insulin sensitivity by euglycemic clamps, assess basal metabolic rate using metabolic cages, and VO2 max by treadmill exercise testing. The ewect of knockouts are also assessed using metabolomic and lipidomic profiles from plasma and liver, as well as RNA sequencing of white adipose tissue (subcutanous and visceral), brown adipose tissue, skeletal muscle and liver.