Software
Bioinformatics algorithms

MethylMix
This DNA methylation is being recognized as an important mechanism that contributes to oncogenesis. Hyper- and hypo-methylation of genes have been identified as critical events in multiple human cancers. A vast amount of data is now available that allows a comprehensive genome wide study of DNA methylation levels of cancer compared to normal tissue. Yet few statistical algorithms exist that exploit these vast datasets to quantify methylation levels and identify hypo- and hyper-methylated genes in cancer. We developed a new computational algorithm called MethylMix based on a Gaussian mixture modeling to identify DNA methylation states relative to normal tissue. Using paired gene expression data, MethylMix also associates DNA methylation with gene expression to identify functional DNA methylation events. We applied MethylMix on ovarian cancer, breast cancer and glioblastoma multiforme data from The Cancer Genome Atlas (TCGA). We identified meaningful hyper- and hypo-methylated genes and differentially methylated pathways in common and distinct to all three human cancers. Our analysis suggests that DNA methylation silences genes that are responsible for transitioning away from a stem-like state for all three cancers. Moreover, we found common genomic locations of methylation in all three cancers; for example, functional differential methylated CpG sites are closer to transcription start sites. Finally, by transforming the methylation datasets based on the mixture components derived from MethylMix, we searched for patient subgroups with similar methylation patterns compare with normal methylation. We identified known subgroups in breast cancer and glioblastoma multiforme, and a new group in ovarian cancer that appears to be driven by a regulatory T cell response.
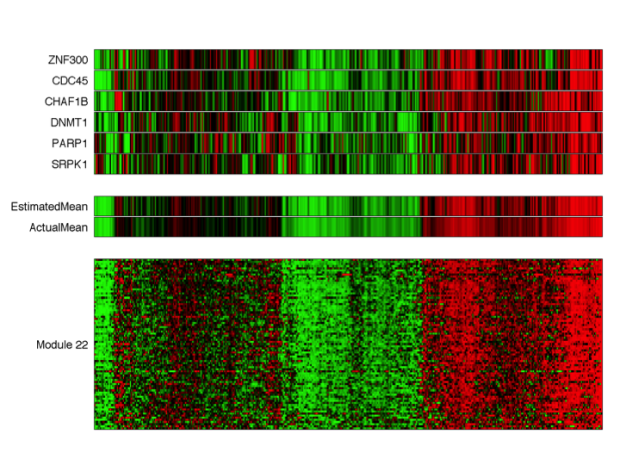
AMARETTO
Integrating an increasing number of available multi-omics cancer data remains one of the main challenges to improve our understanding of cancer. One of the main challenges is using multi-omics data for identifying novel cancer driver genes. We have developed an algorithm, called AMARETTO, that integrates copy number, DNA methylation and gene expression data to identify a set of driver genes by analyzing cancer samples and connects them to clusters of co-expressed genes, which we define as modules. We applied AMARETTO in a pancancer setting to identify cancer driver genes and their modules on multiple cancer sites. AMARETTO captures modules enriched in angiogenesis, cell cycle and EMT, and modules that accurately predict survival and molecular subtypes. This allows AMARETTO to identify novel cancer driver genes directing canonical cancer pathways.
AMARETTO is currently under review at Bioconductor and will be available soon.
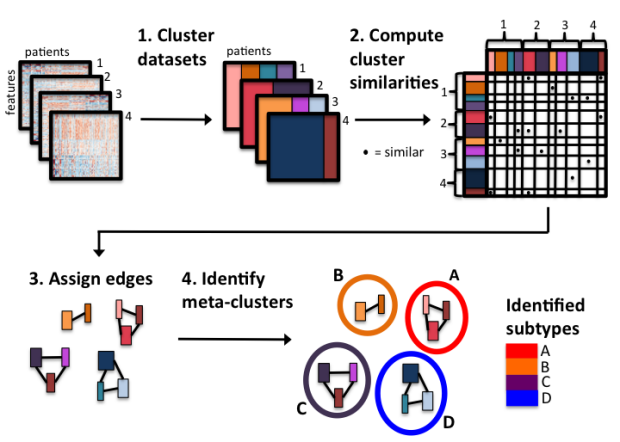
CoINcIDE: Clustering Intra and Inter Datasets
Robust patient clusters derived from gene expression data should be repeatable across multiple datasets. However, there remain few solutions to compare and analyze clustering outputs run on greater than two individual datasets. Current solutions require merging together all datasets; resulting in a loss of dataset-level information. CoINcIDE, Clustering INtra and Inter DatasEts, is a novel method and R package that simultaneously analyzes clustering outputs from each dataset to compute meta-clusters across all datasets. We show that this method discovers repeatable cancer subtypes for both breast and ovarian cancer, and define subtypes that have significant biological and clinical characteristics.
CoINcIDE code is available here.
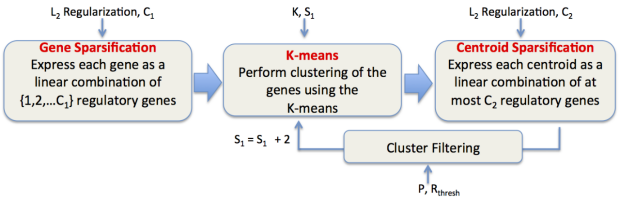
CaMoDi: Cancer Module Discovery
CaMoDi (Cancer Module Discovery) is a fast, scalable computational method that allows users to link potential drug targets with molecular pathways in an accurate and robust manner. CaMoDi uses unsupervised clustering techniques to process larger data sets than other current algorithms. The software can be used to analyze genomic patterns in tumors and predict drug targets to specific diseases and across anatomically defined cancer sites. It is particularly useful analyzing large data sets for identifying “cancer driver” genes.
Metadata prediction
In our recent work on metadata prediction, we describe a framework to predict structured metadata terms from unstructured metadata to improve the quality and quantity of metadata for the GEO microarray database. We developed two approaches for metadata prediction: TF-IDF and Latent Dirichlet Allocation (LDA). We use a Latent Dirichlet Allocation (LDA) model in combination with machine learning methods to effectively predict structured metadata. Our results show that predicting metadata is promising approach `that is likely to be applicable to other datasets and has implications for researchers and practitioners interested in biomedical metadata curation and metadata prediction.
Code to use for metadata prediction can be found here.