Multi-scale data fusion
The main focus of the Gevaert lab is on biomedical data fusion: the development of machine learning methods for biomedical decision support using multi-scale biomedical data. Previously our work was focused on Bayesian and kernel methods studying breast and ovarian cancer. Subsequent work concerned the development of methods for multi-omics data fusion. This resulted in the development of MethylMix, to identify differentially methylation driven genes, and AMARETTO, a computational method to integrate DNA methylation, copy number and gene expression data to identify cancer modules. Additionally, the lab focuses on multi-scale data fusion by linking omics with cellular and tissue-level phenotypes. This led to key contributions in the field of imaging genomics/radiogenomics involving work in lung cancer and brain tumors. The work in imaging genomics is focused on developing a framework for non-invasive personalized medicine. In summary, the Gevaert lab has an interdisciplinary focus on developing novel algorithms for multi-scale biomedical data fusion.
Themes
Multi-scale data fusion
Complex diseases often arise as a multi-scale process involving genetic changes that lead to cellular changes giving rise to lesions at the tissue level, and if left untreated affecting the entire organism. Many studies focus on studying genetic changes and molecular data from genomics technologies such as high throughput sequencing and mass spectrometry. Next, immunohistochemistry (IHC) of cancer tissues provides information of cellular architecture in a visual way for pathologists to interpret. Similarly, radiology images of tumors such as MR images or CT images provide the radiologists visual data of the entire tumor morphology and 3D structure These images reflecting cellular and tissue scale heterogeneity. Together, molecular data, IHC and radiology images potentially contain synergistic information that can help diagnose or treat the patient.
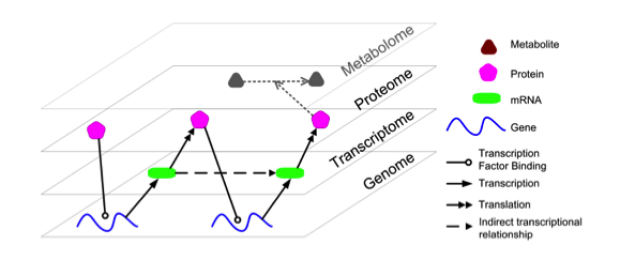
Multi-omics data fusion
The lab has developed multiple frameworks for biomedical data fusion. Initially we developed an extensive framework using Bayesian algorithms. Next, we included algorithms using support vector machines and kernel methods. More recently, we have expanded our work into regularized regression approaches to build data fusion methods for multi-omics data. Most recently we developed AMARETTO and CaMoDi, these are algorithms for multi-omics data fusion. They model gene expression, DNA copy number and DNA methylation data and represent this as cancer modules, and have been shown to outperform existing methods. More recently we are linking multi-omics data across scales with cellular and imaging phenotypes and extend towards multi-scale biomedical data fusion.
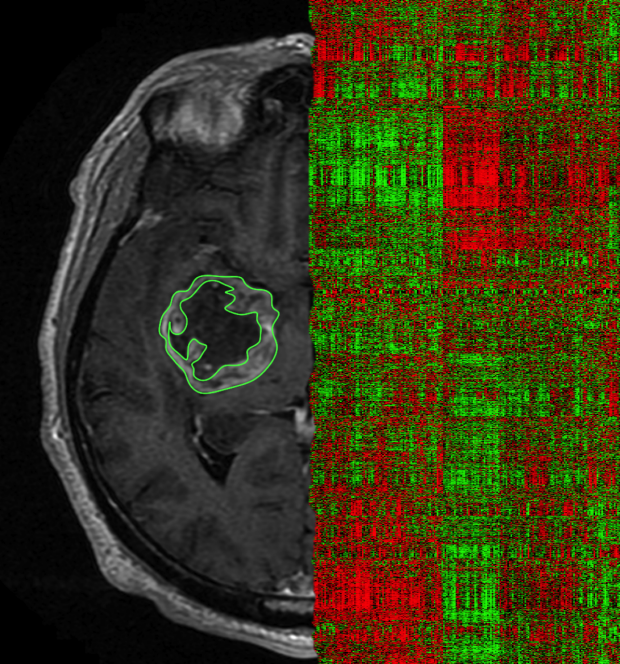
Radiogenomics/Imaging genomics
Molecular profiles of tumors are nowadays used to determine prognosis and to guide therapy. For example the presence of a mutation in the EGFR gene will most likely lead to anti-EGFR therapy. Recently an image phenotype was discovered that acts as a biomarker of EGFR mutation. This is a precursor of the possibilities of a new emerging field called radiogenomics defined as directly linking imaging features to underlying molecular properties. This shows the potential of image signatures that reflect molecular properties of diseases. Especially if these molecular properties are actionable, non-invasive precion medicine becomes a reality.
Read more about an example studying glioblastoma here.
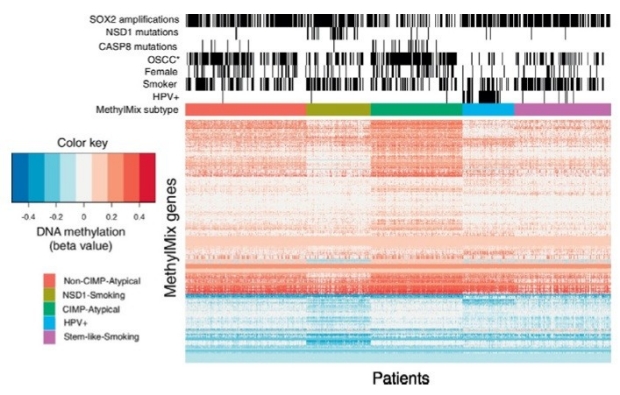
Computational epigenomics
Epigenomic mechanisms such as DNA methylation and histone methylation are important processes known to determine cell type and differentiation state, and are often dysregulated in cancer. The most studied aspect of the epigenome is the DNA methylation landscape, especially in cancer. DNA methylation changes are highly prevalent in every cancer type, occur more frequently than genetic alterations in human cancers and can result in gene expression modulation. Moreover, specific DNA methylation patterns are associated with many common causes of cancer including age, smoking, viruses or bacteria, and DNA methylation also reflects cellular heterogeneity, such as infiltration of lymphocytes cells, macrophages or endothelial cells. Defining cancer subtypes based on DNA methylation changes in cancer tissues vs. normal tissues is thus a promising approach to study cancer etiology.
Projects
Multi-scale model of brain tumors to improve treatment decisions.
Computational multi-scale modeling is a growing area of research that aims to link whole slide images and radiographic iamges with multi-omics molecular profiles of the same patients. Multi-scale modeling has shown its potential through its ability to predict clinical outcomes e.g. prognosis, and through predicting actionable molecular properties of tumors, e.g. the activity of EGFR, a major drug target in many cancers. Current applications are limited to study associations between imaging and molecular data, and predicting long term outcomes. No actionable information can be gained from multi-scale biomarkers yet. We propose to develop a multi-scale modeling framework to support treatment response, treatment monitoring and treatment allocation for patients with brain tumors, focusing on the most aggressive subtype of glioma, IDH wild-type high grade glioma. This project is funded by R01 XXX by the National Cancer Institute (NCI).

Digital twin for cancer patients.
We are developing a Digital Twin (DT) by integrating baseline multi-modal data and repeated measurements for real-time dynamic model training and updating. We will take advantage of innovations in the areas of cancer clinical and biological research, molecular profiling, artificial intelligence, and computing technologies to realize a DT for monitoring treatment response vs. treatment resistance. More specifically, we propose an adaptive dynamic DT to predict response to initial therapy as well as in the maintenance phase to assess resistance mechanisms and enable rapid and effective treatment reassignment. This DT will help physicians make initial treatment determinations, monitor treatment response and effectiveness, and decide when to discontinue or change approach. This work has been funded by the Department of Energy (DoE) & National Cancer Institute (NCI). Read more here.

Organoid-based Discovery of Oncogenic Drivers and Treatment Resistance Mechanisms
Cancer Target Disovery and Development - Stanford Center
We are actively involed in the CTD2 project together with the labs of Calvin Kuo, Christina Curtis and Hanlee Ji. We are developing methods to identify hypomethylated oncogene candidates using novel computational algorithms like MethylMix and extensions to our epigenomic algorithmic work. Oncogenes activated by hypomethylation have been understudied relative to the converse process of gene repression by hypermethylation and represent putative targets for therapeutic inhibition.
More information about the CTD2 network can be found here. All data produced by the network is availbale in raw format here, including our contributions identifying oncogenes using MethylMix.
The CTD2 network has also developed the Dashboard a way to view results assembled across multiple centers for both computational biologistis and those with litle bioinformatics expertise.
Disease focus areas
- Oncology: Brain tumors, lung cancer, Head and neck cancer and Hepatocellular caricinoma
- Cardiovascular diseases
- Neurodegenerative diseases.
Previous research topics
Metadata prediction
Our goal is to study the creation of comprehensive and expressive metadata for biomedical datasets to facilitate data discovery, data interpretation, and data reuse. By facilitating the submission of high quality metadata to describe biomedical experiments, our framework enables experiments to be verified and for disparate data to be integrated. It is imperative to make the authoring of high quality metadata a manageable task. In our work we adopt a data-driven approach for value set recommendation. We used the metadata that investigators have already defined to infer metadata attributes that are commonly used in metadata submission. We use these data to recommend values for the newly entered metadata that need to be filled in during the data submission. Similarly, we use our methods to enhance metadata of existing repositories.
More about our recent results in this area here.