
Biomedical data fusion
The Gevaert lab focuses on biomedical data fusion of complex diseases with a particular focus on oncology and cardiovascular diases. We develop novel machine learning approaches that digest multi-omics, multi-modal or multi-scale data. Previously we pioneered data fusion work using Bayesian and kernel methods studying breast and ovarian cancer. Subsequent work concerned the development of methods for multi-omics data fusion. This resulted in the development of MethylMix, to identify differentially methylated genes, and AMARETTO, a computational method to integrate DNA methylation, copy number and gene expression data to identify cancer modules. Additionally, my lab focuses on linking molecular data with cellular and tissue-level phenotypes. This led to key contributions in the field of imaging genomics/radiogenomics involving work in lung cancer and brain tumors. Our work in imaging genomics is focused on developing a framework for non-invasive personalized medicine. In summary, my lab has an interdisciplinary focus on developing novel algorithms for multi-scale biomedical data fusion.
News
-
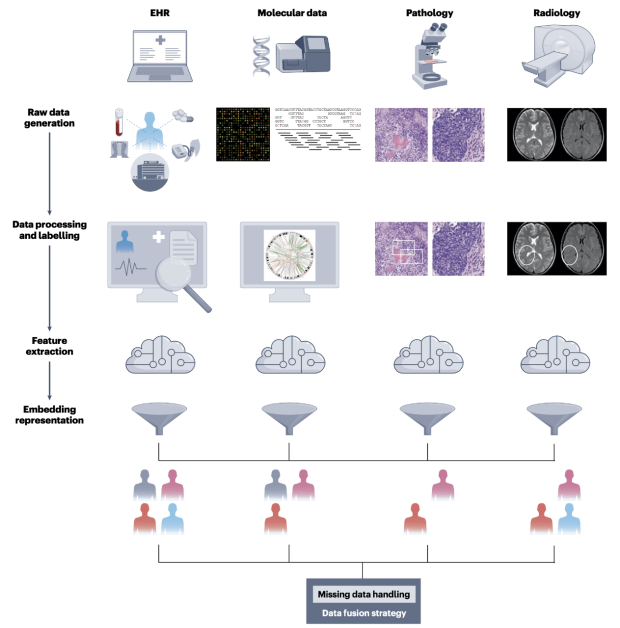
Perspective on multi-modal modeling for biomarker discovery in oncology
Development of effective multimodal fusion approaches is becoming increasingly important as a single modality might not be consistent and sufficient to capture the heterogeneity of complex diseases to tailor medical care and improve personalized medicine.
-

Deep learning for monkeypox
The Gevaert lab spearheaded by visiting scholar Dr. Alexander Thieme has developed a model that is able to distinguish mpox skin lesions from other skin lesions.
-
Imaging genomics: data fusion in uncovering disease heritability
Sequencing of the human genome in the early 2000s enabled probing of the genetic basis of disease on a scale previously unimaginable.
-
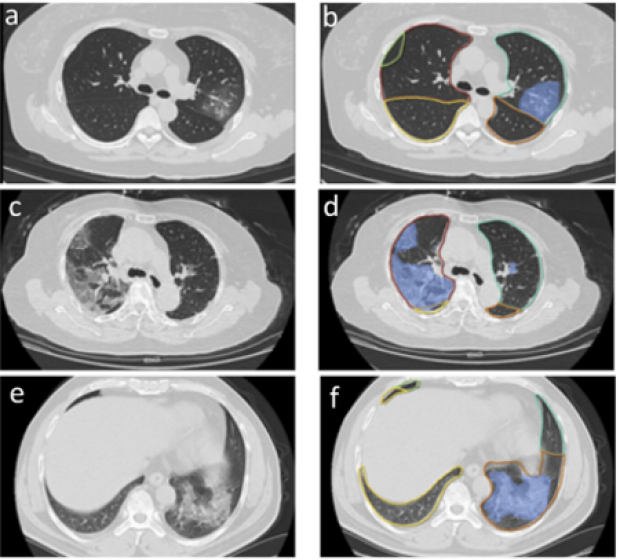
AI based analysis of imaging, clinical & lab data to triage COVID19 patients.
We took advantage of a large cohort of Chinese PCR-confirmed COVID19 patients to embark on a biomedical data fusion project to triage COVID19 patients.
Example projects
We focus on many projects within the space of multi-scale data fusion, here are a few examples:
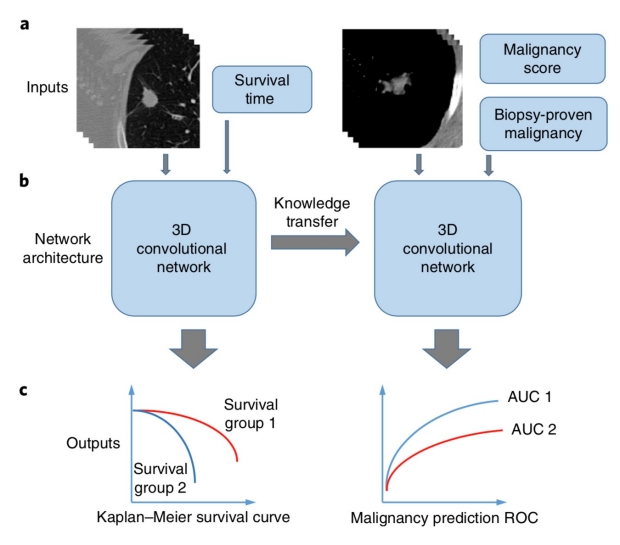
LungNet: Predicting lung cancer survival using CNNs on CT images.
We developed a shallow convolutional neural network (CNN) model and evaluated this on multi-institutional data showing that LungNet outperforms radiomics features and clinical data when predicting overal survival.
Read more about this work that appeared on the cover of Nature Machine Intelligence, Mukerhjee et al. 2020.
Organoid-based Discovery of Oncogenic Drivers and Treatment Resistance Mechanisms
Cancer Target Disovery and Development - Stanford Center
We are actively involed in the CTD2 project together with the labs of Calvin Kuo, Christina Curtis and Hanlee Ji. We are developing methods to identify hypomethylated oncogene candidates using novel computational algorithms like MethylMix and extensions to our epigenomic algorithmic work. Oncogenes activated by hypomethylation have been understudied relative to the converse process of gene repression by hypermethylation and represent putative targets for therapeutic inhibition.
More information about the CTD2 network can be found here. All data produced by the network is availbale in raw format here, including our contributions identifying oncogenes using MethylMix.
The CTD2 network has also developed the Dashboard a way to view results assembled across multiple centers for both computational biologistis and those with litle bioinformatics expertise.

Cancer epigenomics
Aberrant DNA methylation is an important mechanism that contributes to oncogenesis. Yet few algorithms exist that exploit this vast dataset to identify hypo and hypermethylated genes in cancer. We developed a novel computational algorithm called MethylMix to identify differentially methylation genes that are also predictive of transcription. We applied MethylMix on twelve individual cancer sites and combining all cancer sites in a pancancer analysis. We discovered pancancer hyper and hypomethylated genes and identified novel methylation driven subgroups with clinical implications. MethylMix analysis on all cancer sites combined revealed ten pancancer clusters reflecting new similarities across malignantly transformed tissues.
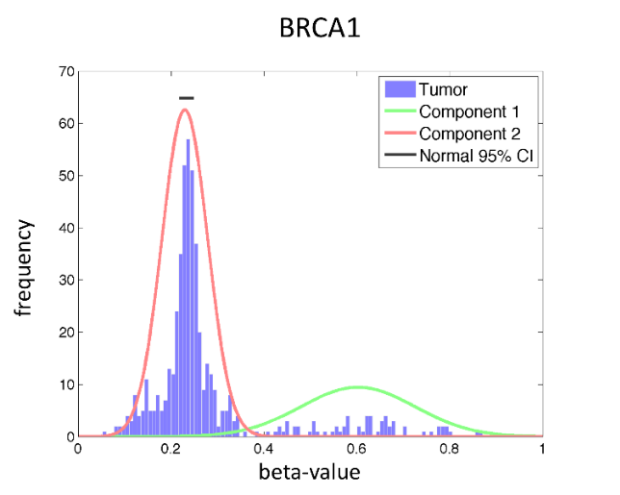
Linking pathology and epigenomics using computational analysis
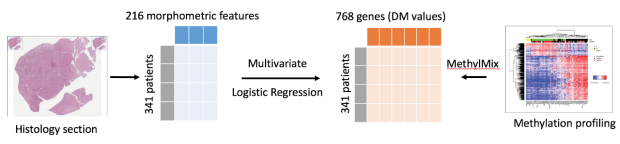
DNA methylation is an important mechanism regulating gene transcription, and its role in carcinogenesis has been extensively studied. Hyper and hypomethylation of genes is a major mechanism of gene expression deregulation in a wide range of diseases. At the same time, high-throughput DNA methylation assays have been developed generating vast amounts of genome wide DNA methylation measurements. However, these assays remain expensive and they are not performed systematically. In parallel, pathologists analyse cancer tissues samples routinely with immuno-histochemistry (IHC).
In this study, we investigate the interactions between computational analysis of IHC images and DNA methylation for a cohort of glioma's including glioblastoma (GBM) and lower grade glioma (LGG). We demonstrate that machine learning algorithms can be used to predict the methylation profile of a patient from morphometric features extracted from IHC images of cancer tissues. This methylation profile spans many genes and we also applied consensus clustering to predict the methylation state of genes clusters. The generalization power of our model was assessed by applying it to an independent type of cancer: Kidney Renal Clear Cell Carcinoma (KIRC).
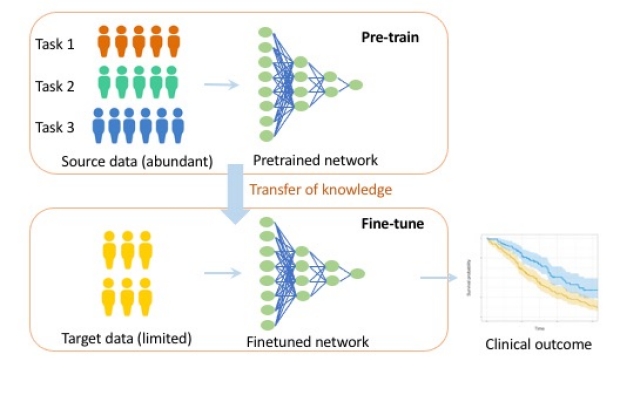
Meta-learning to reduce amount of data needed in AI models
We are actively developing meta-learning approaches to reduce the amount of biomedical data that is needed in a target domain. In recent works, meta learning has shown a lot of promise, in pariculare in studies involving data of cancer patients. Like transfer learning, meta learning takes advantage of huge related data cohorts in a source domain to inform a task in a target domain where data is more limited. In one example, we used meta-learning to predict survival using genomic data of cancer patients and show that it reduces the amount of data needed by one order of magnitude. See: Qiu et al. Nature Communications 2020. We also provide a comment on meta-learning and other success stories in the oncology literature here: Gevaert O. British Journal of Cancer 2021. Read more here.
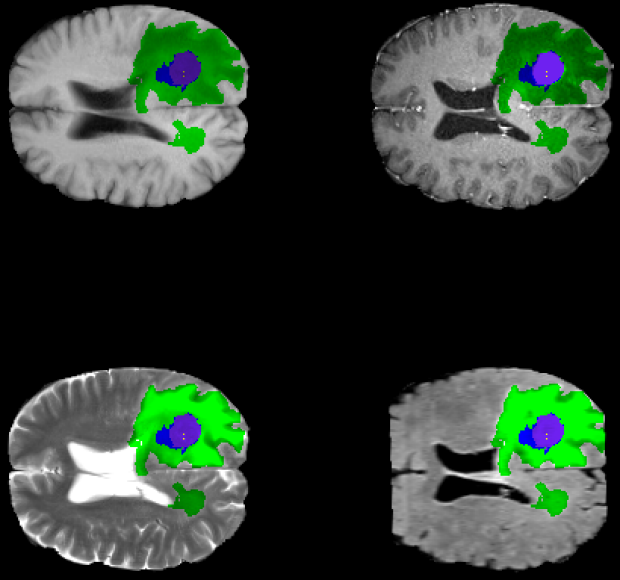
Deep learning for brain tumor segmentation
Improved methods for characterizing tumors both radiologically and histologically are essential for identifying prognostic biomarkers to guide clinical decisions. We developed an algorithm using convolutional neural networks (CNNs) to segment tumors and classify specific regions of interest. By generalizing CNNs to true 3-D convolutions and using a unique architecture to decouple pixels and expand effective data size, our method achieves a median Dice score accuracy of over 90% in whole tumor glioblastoma segmentation, a significant improvement over past algorithms. This result demonstrates the power of our approach in generalizing low-bias methods like CNNs to learn from medium-size medical data sets.
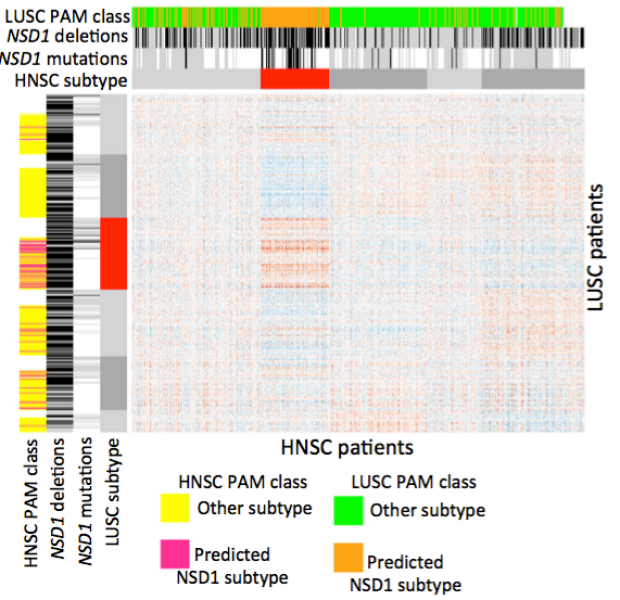
NSD1 inactivation defines an immune cold, DNA hypomethylated subtype in squamous cell carcinoma
Recent reports indicate that inactivating mutations in the histone methyltransferase NSD1 define an intrinsic subtype of head and neck squamous cell carcinoma (HNSC) that features widespread DNA hypomethylation. Here, we describe a similar DNA hypomethylated subtype of lung squamous cell carcinoma (LUSC) that is enriched for both inactivating mutations and deletions in NSD1. The 'NSD1 subtypes' of HNSC and LUSC are highly correlated at the DNA methylation and gene expression levels, with concordant DNA hypomethylation and overexpression of a strongly overlapping set of genes, a subset of which are also hypomethylated in Sotos syndrome, a congenital growth disorder caused by germline NSD1 mutations. Further, the NSD1 subtype of HNSC displays an 'immune cold' phenotype characterized by low infiltration of tumor-associated leukocytes, particularly macrophages and CD8+ T cells, as well as low expression of genes encoding the immunotherapy target PD-1 immune checkpoint receptor and its ligands PD-L1 and PD-L2. Using an in vivo model, we demonstrate that NSD1 inactivation results in a reduction in the degree of T cell infiltration into the tumor microenvironment, implicating NSD1 as a tumor cell-intrinsic driver of an immune cold phenotype. These data have important implications for immunotherapy and reveal a general role of NSD1 in maintaining epigenetic repression.
Pancancer AMARETTO: Multi-omics data fusion to identify cancer driver genes
The availability of increasing volumes of multi-omics profiles promises to improve our understanding of the regulatory mechanisms underlying cancer. The main challenge remain to integrate these multiple levels of omics profiles and especially to analyze them across many cancers. We have developed pancancer AMARETTO, an algorithm that addresses both challenges in three steps. First, AMARETTO identifies potential cancer driver genes through integration of copy number, DNA methylation and gene expression data. Then AMARETTO connects these driver genes with co-expressed target genes that they regulate in regulatory modules. Thirdly, we connect modules identified into a pancancer network to identify cancer driver genes. We applied pancancer AMARETTO on eleven cancer sites and idenditifed pancancer driver genes of smoking-induced cancers and ‘antiviral’ interferon-modulated innate immune response.

Funding

